
Abstract
The discovery of Toll-like receptors (TLRs) as components that recognize conserved structures in pathogens has greatly advanced understanding of how the body senses pathogen invasion, triggers innate immune responses and primes antigen-specific adaptive immunity. Although TLRs are critical for host defense, it has become apparent that loss of negative regulation of TLR signaling, as well as recognition of self molecules by TLRs, are strongly associated with the pathogenesis of inflammatory and autoimmune diseases. Furthermore, it is now clear that the interaction between TLRs and recently identified cytosolic innate immune sensors is crucial for mounting effective immune responses. Here we describe the recent advances that have been made by research into the role of TLR biology in host defense and disease.
Similar content being viewed by others

Main
During the past decade, there has been rapid progress in the understanding of innate immune recognition of microbial components and its critical role in host defense against infection. The early concept of innate immunity was that it nonspecifically recognized microbes; however, the discovery of Toll-like receptors (TLRs) in the mid-1990s showed that pathogen recognition by the innate immune system is instead actually specific, relying on germline-encoded pattern-recognition receptors (PRRs) that have evolved to detect components of foreign pathogens referred to as pathogen-associated molecular patterns (PAMPs)1,2. TLRs are type I transmembrane proteins with ectodomains containing leucine-rich repeats that mediate the recognition of PAMPs; transmembrane domains; and intracellular Toll–interleukin 1 (IL-1) receptor (TIR) domains required for downstream signal transduction. So far, 10 and 12 functional TLRs have been identified in humans and mice, respectively, with TLR1–TLR9 being conserved in both species. Mouse TLR10 is not functional because of a retrovirus insertion, and TLR11, TLR12 and TLR13 have been lost from the human genome. Studies of mice deficient in each TLR have demonstrated that each TLR has a distinct function in terms of PAMP recognition and immune responses3. Elucidation of the crystal structure of several TLR ectodomains has provided structural insights suggesting that several PAMPs act as 'ligands' for TLRs4. PAMPs recognized by TLRs include lipids, lipoproteins, proteins and nucleic acids derived from a wide range of microbes such as bacteria, viruses, parasites and fungi3. The recognition of PAMPs by TLRs occurs in various cellular compartments, including the plasma membrane, endosomes, lysosomes and endolysosomes3. The proper cellular localization of TLRs is thought to be important for ligand accessibility, the maintenance of tolerance to self molecules such as nucleic acids and downstream signal transduction.
TLR signaling pathways were intensively studied after the discovery of the TIR domain–containing adaptor molecule MyD88. The subsequent identification of additional TIR domain–containing adaptors has shown that individual TLRs selectively recruit distinct adaptor molecules, providing specific immunological responses tailored to the infecting microbes3. Studies have indicated that there are cell type–specific signaling pathways that define their immunological properties. For example, plasmacytoid dendritic cells (pDCs) and inflammatory monocytes have unique signaling pathways that govern antiviral responses that are probably absent in other cell types5,6. Recently, much attention in the field of TLR signaling has focused on post-transcriptional modifications, the spatial regulation of signaling molecules and the characterization of TLR target genes.
After the discovery of TLRs, several classes of cytosolic PRRs, including RIG-I-like receptors (RLRs) and Nod-like receptors (NLRs), were identified. The RLR family consists of three members, RIG-I, Mda5 and LGP2, that detect RNA viruses7. The NLR family consists of more than 20 members, and several respond to the various PAMPs, non-PAMP particles and cellular stresses to trigger proinflammatory responses, including the secretion of IL-1β8,9. In addition, cells express as-yet-unidentified PRRs that recognize double-stranded (dsDNA) and induce the production of type I interferon10,11. These PRRs are expressed by many cell types, including nonimmune cells, and in some cases recognize PAMPs shared with TLRs. These PRRs, in concert with TLRs, have a critical role in both innate and adaptive immune responses.
Although TLRs are essential for protective immunity against infection, inappropriate TLR responses contribute to acute and chronic inflammation, as well as to systemic autoimmune diseases. Indeed, mice with defects in the negative regulation of TLR-mediated responses develop these diseases. More importantly, there is growing evidence to indicate that endogenous molecules produced by dying cells, or in certain pathological conditions, stimulate TLRs, resulting in the development or acceleration of inflammatory and autoimmune diseases.
In this Review, we survey the present knowledge of the structural biology, cell biology and signaling of TLRs. We then describe the contributions of TLRs and cytosolic PRRs to adaptive immune responses and, finally, discuss recent progress in TLR-mediated recognition of endogenous molecules and their roles in immune diseases.
Structure and ligands for cell surface TLRs
TLRs are largely divided into two subgroups depending on their cellular localization and respective PAMP ligands. One group is composed of TLR1, TLR2, TLR4, TLR5, TLR6 and TLR11, which are expressed on cell surfaces and recognize mainly microbial membrane components such as lipids, lipoproteins and proteins; the other group is composed of TLR3, TLR7, TLR8 and TLR9, which are expressed exclusively in intracellular vesicles such as the endoplasmic reticulum (ER), endosomes, lysosomes and endolysosomes, where they recognize microbial nucleic acids.
TLR4, a founding member of the TLR family, was identified as the long-sought receptor that responds to bacterial lipopolysaccharide (LPS), a component of the outer membrane of Gram-negative bacteria that can cause septic shock3. TLR4 forms a complex with MD2 on the cell surface, and together they serve as the main LPS-binding component12. A structural study of TLR4-MD2 in complex with LPS has shown that five of the six lipid chains of LPS bind the hydrophobic pocket of MD2, and the remaining lipid chain that is exposed to the surface on MD2 associates with TLR4 (refs. 13,14; Fig. 1). The phosphate groups also interact with the positively charged residues of TLR4. The resultant formation of a receptor multimer composed of two copies of the TLR4-MD2-LPS complex initiates signal transduction by recruiting intracellular adaptor molecules. Additional proteins such as LPS-binding protein (LBP) and CD14 are also involved in LPS binding12. LBP is a soluble plasma protein that binds LPS, and CD14 is a glycosylphosphatidylinositol-linked, leucine-rich repeat–containing protein that binds LBP and delivers LPS-LBP to the TLR4-MD2 complex. In addition to binding LPS, TLR4 is involved in the recognition of respiratory syncytial virus fusion proteins, mouse mammary tumor virus envelope proteins, Streptococcus pneumoniae pneumolysin and the plant-derived cytostatic drug paclitaxel3, although structural insights into the interaction between TLR4 and these ligands have not yet been provided.

TLR4 in complex with MD2 engages LPS. Five of the six lipid chains of LPS bind MD2 and the remaining lipid chain associates with TLR4. The formation of a receptor multimer composed of two copies of the TLR4-MD2-LPS complex initially transmits signals for the early-phase activation of NF-κB by recruiting the TIR domain–containing adaptors TIRAP (Mal) and MyD88 (MyD88-dependent pathway). The TLR4-MD2-LPS complex is then internalized and retained in the endosome, where it triggers signal transduction by recruiting TRAM and TRIF, which leads to the activation of IRF3 and late-phase NF-κB for the induction of type I interferon (TRIF-dependent pathway). Both early- and late-phase activation of NF-κB is required for the induction of inflammatory cytokines. TLR2-TLR1 and TLR2-TLR6 heterodimers recognize triacylated and diacylated lipopeptide, respectively. Two of the three lipid chains of the triacylated lipopeptide interact with TLR2, and the third chain binds the hydrophobic channel of TLR1 (absent from TLR6). TLR2-TLR1 and TLR2-TLR6 induce NF-κB activation through recruitment of TIRAP and MyD88. TLR5 recognizes flagellin and activates NF-κB through MyD88.
TLR2 is involved in the recognition of a wide range of PAMPs derived from bacteria, fungi, parasites and viruses3. These include lipopeptides from bacteria, peptidoglycan and lipoteichoic acid from Gram-positive bacteria, lipoarabinomannan from mycobacteria, zymosan from fungi, tGPI-mucin from Trypanosoma cruzi and the hemagglutinin protein from measles virus. TLR2 generally forms heterodimers with TLR1 or TLR6. Specifically, the TLR2-TLR1 heterodimer recognizes triacylated lipopeptides from Gram-negative bacteria and mycoplasma, whereas the TLR2-TLR6 heterodimer recognizes diacylated lipopeptides from Gram-positive bacteria and mycoplasma. Studies have provided structural insights into the mechanisms by which these heterodimers discriminate the structures of lipoproteins15,16. TLR2-TLR1 and TLR2-TLR6 heterodimers share an m-shaped structure (Fig. 1). In the TLR2-TLR1–ligand complex, two of the three lipid chains of Pam3CSK4 (a triacylated lipopeptide) interact with TLR2, whereas the third chain binds the hydrophobic channel of TLR1. Thus, recognition of the triacylated lipopeptide is facilitated. However, the hydrophobic channel is absent from TLR6, so the TLR2-TLR6 heterodimer does not recognize triacylated lipopeptides. Together, the different lipid-binding pockets of TLR1 and TLR6 are responsible for the discrimination between lipoproteins. Moreover, TLR2 has the ability to act together with other coreceptors on the cell surface that assist PAMP recognition. These include CD36, which acts together with the TLR2-TLR6 heterodimer to mediate the sensing of some but not all TLR2 agonists17, and dectin-1, a C-type lectin that binds fungus β-glucan and induces its internalization18. Although it was believed that TLR2 agonists induce mainly the production of inflammatory cytokines and not type I interferon by macrophages and dendritic cells (DCs), it can trigger the production of type I interferon by inflammatory monocytes in response to infection with vaccinia virus6, which suggests a cell type–specific role for TLR2 in antiviral responses. It seems that nucleic acids, which are usually the trigger for the production of type I interferon, do not participate in the activation of TLR2.
TLR5 recognizes the flagellin protein component of bacterial flagella3 (Fig. 1). CD11c+CD11b+ lamina propria DCs in the small intestine have high TLR5 expression19. Lamina propria DCs are unique in promoting the differentiation of IL-17-producing helper T cells (TH17 cells) and T helper type 1 (TH1) cells, as well as the differentiation of naive B cells into immunoglobulin A–producing plasma cells in response to flagellin19. Furthermore, lamina propria DCs are able to produce retinoic acids, which facilitate these humoral and cellular immune responses. The kidney and bladder have high expression of mouse TLR11, which is a relative of TLR5. TLR11 is thought to recognize uropathogenic bacterial components, as TLR11-deficient mice are susceptible to infection with these bacteria20. TLR11 also recognizes the profilin-like molecule derived from Toxoplasma gondii21.
Structure and ligands for nucleic acid–sensing TLRs
TLR3 was originally identified as recognizing a synthetic analog of double-stranded RNA (dsRNA), polyinosinic-polycytidylic acid (poly(I:C)), which mimics viral infection and induces antiviral immune responses by promoting the production of both type I interferon and inflammatory cytokines. The recognition mechanism was elucidated by structural analysis of the human TLR3 ectodomain bound to dsRNA22,23 (Fig. 2). The TLR3 ectodomain has a large horseshoe-like shape that probably functions to increase its surface area and facilitate dsRNA recognition. The dsRNA binds to two different sites at the N and C termini on the lateral side of the convex surface of the TLR3 ectodomain, which provides enough stability to allow TLR3 to form a homodimer via the C-terminal region. In addition to recognizing poly(I:C), TLR3 recognizes the genomic RNA of reoviruses, dsRNA produced during the replication of single-stranded RNA (ssRNA), viruses, including respiratory syncytial virus, encephalomyocarditis virus and West Nile virus, and certain small interfering RNAs3,24. TLR3 triggers antiviral immune responses through the production of type I interferon and inflammatory cytokines, which suggests that TLR3 has an essential role in preventing virus infection. Consistent with that, TLR3-deficient mice are susceptible to lethal infection with murine cytomegalovirus25, and TLR3 deficiency in humans is associated with susceptibility to herpes simplex virus type 1 (HSV-1)26.

TLR3 recognizes dsRNA derived from viruses or virus-infected cells; dsRNA binds to N- and C-terminal sites on the lateral side of the convex surface of the TLR3 ectodomain, which facilitates the formation of a homodimer via the C-terminal region. TLR3 activates the TRIF-dependent pathway to induce type I interferon and inflammatory cytokines. In pDCs, TLR7 recognizes ssRNA derived from ssRNA viruses in endolysosomes and activates NF-κB and IRF7 via MyD88 to induce inflammatory cytokines and type I interferon, respectively. In addition, autophagy is involved in delivering ssRNA to TLR7-expressing vesicles. TLR9 recognizes DNA derived from both DNA viruses and bacteria. Proteolytic cleavage of TLR9 by cellular proteases is required for downstream signal transduction. TLR9 recruits MyD88 to activate NF-κB and IRF7 in pDCs. TLR3, TLR7 and TLR9 localize mainly to the ER in the steady state and traffic to the endolysosomes, where they engage with their ligands. UNC93B1 interacts with these TLRs in the ER and is essential for this trafficking.
TLR7, originally identified as recognizing imidazoquinoline derivatives such as imiquimod and resiquimod (R-848) and guanine analogs such as loxoribine (which have antiviral and antitumor properties), recognizes ssRNA derived from RNA viruses such as vesicular stomatitis virus, influenza A virus and human immunodeficiency virus3,5 (Fig. 2). TLR7 also recognizes synthetic poly(U) RNA and certain small interfering RNAs27. There is high expression of TLR7 on pDCs that are able to produce large amounts of type I interferon after virus infection, and cytokine induction by pDCs in response to RNA viruses is totally dependent on TLR7 (refs. 3,5), which suggests that TLR7 serves as the sensor of infection with ssRNA viruses. Moreover, TLR7 expressed on conventional DCs (cDCs) senses RNA species from bacteria such as group B Streptococcus and induces type I interferon28.
TLR7-mediated recognition of RNA viruses by pDCs occurs in a replication-independent manner. Viruses are internalized and recruited to the endolysosomes, where TLR7-mediated recognition of ssRNA and the initiation of antiviral responses are triggered. Moreover, TLR7 also senses replicating vesicular stomatitis virus that enters the cytoplasm via autophagy, a process for the lysosomal degradation of cellular proteins that involves the formation of double-membrane vesicles called autophagosomes (Fig. 2). The pDCs that lack autophagy-related protein Atg5, which fail to induce autophagosome formation, show defects in the production of interferon-α after infection with vesicular stomatitis virus29. Moreover, pDCs show constitutive autophagosome formation. These findings suggest that pDC autophagy is important for the delivery of cytosolic viral replication intermediates to the lysosome, where TLR7 participates in their recognition and the subsequent initiation of antiviral responses.
TLR8 is phylogenetically most similar to TLR7. Human TLR8 mediates the recognition of R-848 and viral ssRNA. In contrast to mice that lack TLR7, mice that lack TLR8 respond normally to these agonists3,5. TLR8 is expressed in various tissues, with its highest expression in monocytes, and is upregulated after bacterial infection.
TLR9 recognizes unmethylated 2′-deoxyribo(cytidine-phosphate-guanosine) (CpG) DNA motifs that are frequently present in bacteria and viruses but are rare in mammalian cells (Fig. 2). Synthetic CpG oligodeoxynucleotides function as TLR9 ligands and directly activate DCs, macrophages and B cells, and drive strong TH1 responses3. The sugar backbone of DNA, 2′-deoxyribose, is important for TLR9 recognition when the DNA oligonucleotide has a phosphodiester backbone. In contrast, the CpG motif is indispensable in the case of unnatural phosphorothionate backbones30. There is high expression of TLR9 by pDCs, and it serves as a sensor of DNA virus infection (for example, murine cytomegalovirus, HSV-1 and HSV-2)3,5. In addition to recognizing DNA, TLR9 directly recognizes the insoluble crystal hemozoin, which is generated as a byproduct of the detoxification process after digestion of host hemoglobin by Plasmodium falciparum31.
Cellular localization of nucleic acid–sensing TLRs
As mentioned above, nucleic acid–sensing TLRs localize to various intracellular compartments. The finding that blockade of endolysosome acidification prevents TLR7- and TLR9-induced responses suggests that the delivery of internalized nucleic acids to the endolysosomes is pivotal to interaction with these TLRs. TLR9 and TLR7 are exclusively sequestered in the ER in unstimulated cells and rapidly traffic to endolysosomes after ligand stimulation32 (Fig. 2). This translocation is regulated by the ER-localizing protein UNC93B1, a 12 membrane–spanning protein (Fig. 2). Triple D (3d) mice bearing a single missense mutation in the gene encoding UNC93B1 have defects in cytokine production and upregulation of costimulatory molecules in response to TLR7 and TLR9 ligands, as well as TLR3 ligands, and are highly susceptible to viral and bacterial infection33. It is reported that UNC93B1 deficiency is responsible for HSV-1 encephalitis in human patients34. Cells derived from these patients are hyporesponsive to agonists for TLR3, TLR7 and TLR9 but have intact responses to the agonists for other extracellular TLRs. UNC93B1 specifically binds to the transmembrane regions of TLR3, TLR7 and TLR9 in the ER, and TLR7 and TLR9 do not exit the ER in DCs with the 3d mutation32,35. Together these results suggest that UNC93B1 assists in the delivery of both TLR7 and TLR9 from the ER to the endolysosome, which is a prerequisite for the induction of immune responses by these TLRs. Trafficking of TLRs is also regulated by two other proteins residing in the ER: PRAT4A and gp96. PRAT4A associates with TLR4 and TLR9 and is required for the trafficking of TLR4 and TLR9 to the plasma membrane and endolysosme, respectively36. Responses to agonists for TLR2, TLR4 and TLR9 are abrogated in PRAT4A-deficient cells, whereas TLR3-mediated responses are intact in these cells37, which indicates that the trafficking of TLR3 and TLR9 is regulated differently. Macrophages deficient in gp96, a member of the ER-resident heat-shock protein 90 family, have defects in cytokine induction in response to agonists for TLR1, TLR2, TLR4, TLR5, TLR7 and TLR9. Furthermore, gp96 has been shown to bind TLR9. These findings suggest that gp96 functions as a general chaperone for most TLRs, unlike PRAT4, which regulates trafficking of certain TLRs only38.
TLR9 is known to be proteolytically cleaved by intracellular proteases in endolysosomes, which generates a functional receptor that mediates ligand recognition and initiates signal transduction (Fig. 2). The proteases that potentially mediate TLR9 cleavage include cathepsins, such as cathepsin B, cathepsin S, cathepsin L, cathepsin H and cathepsin K, and asparaginyl endopeptidase39,40,41,42,43. However, controversy still exists as to the functional cleavage of TLR9. The deletion of specific leucine-rich repeats in the N-terminal region renders TLR9 unresponsive to its ligand, and the positively charged N-terminal region of TLR9 is proposed to mediate binding to CpG DNA, which suggests the importance of the full-length structure in TLR9 activation44.
TIR domain–containing adaptors in TLR signaling
Individual TLRs trigger specific biological responses. For example, TLR3 and TLR4 generate both type I interferon and inflammatory cytokine responses, whereas cell surface TLR1-TLR2, TLR2-TLR6 and TLR5 induce mainly inflammatory cytokines (Figs. 1 and 2). These differences are explained by the discovery of TIR domain–containing adaptor molecules, including MyD88, TIRAP (Mal), TRIF and TRAM, which are recruited by distinct TLRs and activate distinct signaling pathways (Figs. 1 and 2). MyD88, the first identified member of this TIR family, is universally used by all TLRs except TLR3, and activates the transcription factor NF-κB and mitogen-activated protein kinases (MAPKs) to induce inflammatory cytokines3. In contrast, TRIF is used by TLR3 and TLR4 and induces alternative pathways that lead to activation of the transcription factors IRF3 and NF-κB and the consequent induction of type I interferon and inflammatory cytokines. TRAM and TIRAP function as sorting adaptors that recruit TRIF to TLR4 and MyD88 to TLR2 and TLR4, respectively. Thus, TLR signaling pathways can be largely classified as either MyD88-dependent pathways, which drive the induction of inflammatory cytokines, or TRIF-dependent pathways, which are responsible for the induction of type I interferon as well as inflammatory cytokines3.
TLR4 is the only TLR that uses all four adaptors and activates both the MyD88- and TRIF-dependent pathways (Fig. 1). TLR4 initially recruits TIRAP at the plasma membrane and subsequently facilitates the recruitment of MyD88 to trigger the initial activation of NF-κB and MAPK45. TLR4 subsequently undergoes dynamin-dependent endocytosis and is trafficked to the endosome, where it forms a signaling complex with TRAM and TRIF, rather than TIRAP and MyD88, to initiate the TRIF-dependent pathway that leads to IRF3 activation as well as the late-phase activation of NF-κB and MAPK46,47,48. Thus, TLR4 activates the MyD88-dependent pathway earlier than the TRIF-dependent pathway. Notably, activation of both the MyD88- and TRIF-dependent pathways is necessary for the induction of inflammatory cytokines via TLR4 signaling, which is in contrast to other TLRs, for which activation of either the MyD88- or the TRIF-dependent pathway is sufficient for the induction of inflammatory cytokines. It is still a mystery why activation of either pathway alone is insufficient for the induction of inflammatory cytokines via TLR4 signaling.
The MyD88-dependent pathway
After the engagement of TLRs by their cognate PAMPs, MyD88 recruits the IL-1 receptor–associated kinases IRAK4, IRAK1, IRAK2 and IRAK-M (Fig. 3). IRAK4 is activated initially and has an essential role in the activation of NF-κB and MAPK downstream of MyD88 (ref. 3). IRAK1 and IRAK2 are activated sequentially, and activation of both kinases is required for robust activation of NF-κB and MAPK49. IRAK activation results in an interaction with TRAF6, an E3 ligase that catalyzes the synthesis of polyubiquitin linked to Lys63 (K63) on target proteins, including TRAF6 itself and IRAK1, in conjunction with the dimeric E2 ubiquitin-conjugating enzymes Ubc13 and Uev1A. The K63-linked polyubiquitin chains then bind to the novel zinc finger–type ubiquitin-binding domain of TAB2 and TAB3, the regulatory components of the kinase TAK1 complex, to activate TAK1. The K63-linked polyubiquitin chains also bind to the ubiquitin-binding domain of NEMO, an essential regulatory component of the IKK complex required for NF-κB activation. Thus, the K63 polyubiquitin chains might be responsible for recruiting TAK1 to form a complex with IKK, thus allowing TAK1 to phosphorylate IKKβ through its close proximity to the IKK complex, which leads to NF-κB activation via phosphorylation and subsequent degradation of IκB proteins50. However, Ubc13-deficient cells show intact NF-κB activation in response to TLR agonists, despite the defective K63-linked polyubiquitination of NEMO, which suggests that a K63-linked ubiquitination-independent mechanism exists in NEMO-mediated NF-κB activation51. Head-to-tail linear polyubiquitination of NEMO by the linear ubiquitin-chain assembly complex (which consists of HOIL-1L and HOIP) has been shown to be an important process for IKK activation52,53,54.

TLR-mediated responses are controlled mainly by the MyD88-dependent pathway, which is used by all TLRs except TLR3, and the TRIF-dependent pathway, which is used by TLR3 and TLR4. TRAM and TIRAP are sorting adaptors used by TLR4 and TLR2-TLR4, respectively. In cDCs and macrophages, MyD88 recruits IRAK4, IRAK1, IRAK2 and TRAF6 and induces inflammatory responses by activating NF-κB, MAPK and IRF5. TRAF6 activates TAK1 in complex with TAB2 and TAB3 and activates the IKK complex consisting of NEMO and IKKαβ, which catalyze IκB proteins for phosphorylation. NF-κB induces C/EBPδ, IκBζ, IκB-NS, Zc3h12a, ATF3 and tristeraprolin (TTP), which influence the genes encoding IL-6, IL-12p40 or TNF. TRIF recruits TRAF6, TRADD and TRAF3. TRADD interacts with Pellino-1 and RIP1. RIP1 and TRAF6 cooperatively activate TAK1, which leads to activation of MAPK and NF-κB. TRAF3 activates the kinases TBK1 and IKKi, which phosphorylate and activate IRF3, the last of which controls transcription of type I interferon. Nrdp1 is involved in TBK1-IKKi activation. The TRIF-dependent pathway leads to inflammasome activation during TLR4 signaling. In pDCs, TLR7 and TLR9 recruit MyD88 along with IRAK4 and TRAF6, which activate IRF5 and NF-κB for inflammatory cytokine induction and IRF7 for type I interferon induction. For IRF7 activation, IRAK1- and IKKα-dependent phosphorylation is required, and TRAF3 is located upstream of these kinases. OPNi is involved in IRF7 activation, and IRF8 facilitates NF-κB activation. The PI(3)K-mTOR-p70S6K axis enhances the TLR7 and TLR9 signaling pathways. IRF1 is involved in the induction of type I interferon by TLR7 and TLR9 in cDCs rather than pDCs. Among the many negative regulators of TLRs that have been identified, TANK (which suppresses TRAF6), A20 (which suppresses TRAF6 and RIP1), ATG16A (which suppresses inflammasome activation) and SHP-1 (which suppresses IRAK1 and IRAK2) are reported to be indispensable for preventing inflammatory diseases caused by enhanced or prolonged TLR signaling. Yellow, TLRs; green, stimulators; pink, negative regulators; blue, target genes.
TAK1 simultaneously activates the MAPKs Erk1, Erk2, p38 and Jnk by inducing the phosphorylation (rather than ubiquitination) of MAPK kinases, which then activate various transcription factors, including AP-1, as well as influencing translation. Despite having normal NF-κB activation, Ubc13-deficient cells show impaired MAPK activation51. However, the direct target of Ubc13 that is responsible for MAPK activation remains unidentified.
Activation of the MyD88-dependent pathway results in the induction of many genes, and some of these have critical roles in modulating NF-κB-dependent transcription (Fig. 3). These include the IκB protein IκBζ, which functions as an inducible coactivator for the NF-κB p50 subunit to facilitate IL-6 and IL-12p40 induction55; C/EBPδ, which acts together with NF-κB to maximize IL-6 production56; IκB-NS, which suppresses the induction of both IL-6 and tumor necrosis factor (TNF) by modulating the DNA-binding activity of the NF-κB p65 subunit57; and ATF3, which restricts NF-κB activity by recruiting histone deacetylase58.
The TRIF-dependent pathway
The TRIF-dependent pathway culminates in the activation of both IRF3 and NF-κB24 (Fig. 3). TRIF recruits TRAF6 and activates TAK1 for NF-κB activation, most probably through ubiquitination-dependent mechanisms similar to those of the MyD88-dependent pathway. TRIF also recruits the adaptor RIP1 through the distinct RIP homotypic interaction motif. RIP1 undergoes K63-linked polyubiquitination after stimulation by TLR3 agonists, and this modification is required for NF-κB activation. The adaptor TRADD binds RIP1, and TRADD-deficient cells show impaired RIP1 ubiquitination with concomitant loss of NF-κB activation59,60, which suggests involvement of TRADD in RIP1 activation downstream of TLR3. Pellino-1 is a member of the Pellino family of RING-like domain–containing E3 ubiquitin ligases, and Pellino-1 deficiency causes loss of RIP1 ubiquitination and NF-κB activation in response to TLR3 agonists, despite normal MyD88-dependent NF-κB activation61. Collectively, TRIF forms a multiprotein signaling complex along with TRAF6, TRADD, Pellino-1 and RIP1 for the activation of TAK1, which in turn activates the NF-κB and MAPK pathways.
In addition to leading to NF-κB activation, the TRIF-dependent pathway leads to IRF3 activation and interferon-β transcription (Fig. 3). TRIF recruits a signaling complex involving the noncanonical IKKs TBK1 and IKKi (IKKɛ), which catalyze the phosphorylation of IRF3 and induce its nuclear translocation62. The activation of TBK1-IKKi by TRIF requires TRAF3. TRAF3 deficiency impairs interferon-β induction by TLR3, as well as by TLR7, TLR9 and RLRs, which indicates a general role for TRAF3 in interferon-β induction by various nucleic acid–sensing PRRs63,64.
TRAF3 is also incorporated into the MyD88 complex during TLR4 signaling. However, this exposes TRAF3 to K48-linked ubiquitination and degradation via cIAP1 and cIAP2, which are both components of the MyD88 signaling complex but not the TRIF signaling complex. TRAF3 degradation results in translocation of the membrane-proximal signaling complex to the cytoplasm, which leads to TAK1 activation65. These findings suggest that TRAF3 promotes IRF3 activation as well as inhibiting the MyD88-dependent pathway. The differences in regulation of the MyD88- and TRIF-dependent pathways by a single molecule has also been reported in studies of NRDP1, a RING-containing E3 ligase that interacts with and potentiates TBK1 activation through K63-linked ubiquitination, for which Ubc13 is required66. At the same time, it inhibits activation of the MyD88-dependent pathway through its interaction with and degradation of MyD88. Balanced production of inflammatory cytokines and type I interferon by these molecules might have key roles in controlling tumor cell growth and autoimmune diseases.
TLR7 and TLR9 signaling in pDCs
The TLR7 and TLR9 signaling pathways in pDCs have been extensively investigated to elucidate their potential to induce the production of type I interferon after viral infection. The TLR7 and TLR9 signaling pathways in pDCs are unique in that they both require MyD88 for the induction of type I interferon (Fig. 3). In this context, IRF7, which is constitutively expressed by pDCs, binds MyD88 and forms a multiprotein signaling complex with IRAK4, TRAF6, TRAF3, IRAK1 and IKKα24 (Fig. 3). In this complex, IRF7 becomes phosphorylated by IRAK1 and/or IKKα, dissociates from the complex and translocates into the nucleus. In addition to requiring phosphorylation, IRF7 activation probably requires TRAF6- and Ubc13-dependent ubiquitination. Whereas IRAK1, IKKα and TRAF3 are specifically involved in the activation of IRF7, MyD88, IRAK4 and TRAF6 are critical for the activation of both IRF7 and NF-κB.
Additional components that control the production of type I interferon by pDCs have also been identified (Fig. 3). A precursor of osteopontin, OPNi is a TLR9-inducible protein that is sequestered in the cytoplasm and functions as a component of the MyD88-IRF7 complex in pDCs67. Pharmacological inhibition of phosphoinositol 3-OH kinase (PI(3)K) abrogates the nuclear translocation of IRF7. Moreover, inhibition of mTOR and p70S6K, both of which are downstream targets of PI(3)K, disrupts the interaction between TLR9 and MyD88, which results in impaired nuclear translocation of IRF7 and induction of type I interferon68. These findings suggest that the PI(3)K-mTOR pathway accelerates the production of type I interferon by pDCs during viral infection. IRF5 is incorporated into the MyD88 complex and controls the induction of IL-6 and IL-12p40 (ref. 69). The MyD88-IRF5 pathway is also used by many TLRs in other cell types, such as macrophages and cDCs. IRF8-null pDCs show loss of TLR9-mediated induction of both type I interferon and inflammatory cytokines, with less NF-κB activation, which suggests the possibility that IRF8, in cooperation with NF-κB, controls cytokine induction70.
Retention of the CpG-TLR9 signaling complex in the endosome is also an important mechanism used by pDCs to control antiviral innate immune responses. The A (D)-type CpG oligodeoxynucleotide, which contains a single CpG motif and a poly(G) tail on a phosphorothioate-phosphodiester backbone, is able to induce the secretion of type I interferon by pDCs. It is stably retained in the early endosomes in pDCs along with TLR9, MyD88 and IRF7 (ref. 71). In contrast, the B (K)-type CpG oligodeoxynucleotide, which contains multiple CpG motifs on a phosphothioate backbone and can induce both IL-12 production by cDCs and B cell activation, is rapidly transferred to late endosomes or lysosomes, which results in less activation of IRF7.
Negative regulators of TLR signaling
The negative regulation of TLR-induced responses is important for suppressing inflammation and deleterious immune responses. So far, many negative regulators that suppress TLR signaling pathways at multiple levels have been identified. These include splice variants for adaptors or their related proteins72,73, ubiquitin ligases74,75, deubiquitinases76, transcriptional regulators and microRNAs77,78. Here we focus on the negative regulators of TLR-mediated immune responses, whose disruption or mutation results in persistent inflammation in vivo.
TANK binds TBK1 and IKKi and has been linked to the activation of both NF-κB and IRF3. TANK-deficient mice spontaneously develop autoimmune glomerular nephritis, which is suppressed by treatment with antibiotics or deficiency in MyD88 or IL-6 (ref. 79). Despite having intact induction of type I interferon, TANK-deficient macrophages and B cells show more NF-κB activation and IL-6 production in response to TLR ligands. TANK-deficient cells also show enhanced TRAF6 ubiquitination. Therefore, TANK acts as a negative regulator of TRAF6 ubiquitination in both macrophages and B cells (Fig. 3).
Mutations in the gene encoding the autophagy-related molecule Atg16L1 have been linked to Crohn's disease80. Atg16L1-deficient mice are highly susceptible to dextran sulfate sodium–induced acute colitis, which is blocked by treatment with antibodies to IL-1β and IL-18 (ref. 81). Moreover, macrophages derived from these mice show more activation of caspase-1 and production of IL-1β and IL-18 in response to LPS. Overactivation of caspase-1 by LPS in the absence of Atg16L1 requires TRIF, which suggests that Atg16L1 negatively regulates the TRIF-dependent pathways that lead to caspase-1 activation (Fig. 3). Moreover, intestinal Paneth cells derived from Atg16L1-deficient mice show higher expression of genes involved in responses to intestinal injury82. Atg16L1 is thus essential for the suppression of intestinal inflammation.
TLR stimulation rapidly induces the regulatory protein Zc3h12a, which contains a CCCH-type zinc-finger domain and an RNase domain. Zc3h12a targets the 3′ untranslated regions of IL-6 mRNA and IL-12p40 mRNA for degradation via its RNase activity. Zc3h12a-deficient macrophages consistently produce remarkably large amounts of IL-6 and IL-12p40 but normal amounts of TNF in response to TLR agonists, and mice deficient in Zc3h112a have higher serum immunoglobulin levels and autoantibody production83. Together these results suggest that Zc3h12a negatively regulates TLR-induced inflammatory responses by affecting mRNA stability and prevents autoimmunity (Fig. 3). Another zinc-finger protein, tristeraprolin (Xfp36), prevents the development of autoimmune arthritis. Tristeraprolin binds AU-rich elements in the 3′ untranslated region of TNF mRNA and removes the poly(A) tail by deadenylation, which leads to degradation84 (Fig. 3). Therefore, these zinc-finger proteins control mRNA stability through different mechanisms.
A20 is a protein induced during TLR stimulation that has two enzymatic activities, acting as an E3 ubiquitin ligase and a deubiquitinase. In vitro analyses have shown that A20 restricts NF-κB activation by modulating RIP1 and TRAF6 (Fig. 3). A20-deficient mice die prematurely due to the spontaneous development of multiorgan inflammation and severe cachexia, which indicates that A20 has anti-inflammatory properties in vivo. Deficiency in both A20 and MyD88 rescues the mice from premature death and diminishes the inflammation in these mice85. Also, the administration of antibiotics prevents cachexia. Thus, A20 might inhibit TLR signaling induced by commensal bacteria.
Mice bearing mutations in the gene encoding the tyrosine phosphatase SHP-1 develop inflammatory lesions associated with aberrant macrophage activation in response to TLR stimulation86. MyD88 deficiency suppresses this inflammation, which suggests that SHP-1 negatively regulates the MyD88-dependent pathway. It has also been reported that SHP-1 suppresses the function of both IRAK1 and IRAK2 (ref. 87; Fig. 3).
Non-TLR cytosolic PRRs in PAMP recognition
The innate immune system recognizes cytoplasmic PAMPs through RLRs, NLRs and an unidentified dsDNA sensor. RLRs (RIG-I, Mda5 and LGP2) are RNA helicases that detect viral RNA species and signal through the adaptor molecule IPS-1, thus inducing antiviral responses7,24. NLRs represent a large family of PRRs that respond to various stimuli, including PAMPs, non-PAMP particles and cellular stresses8,9. Among the NLRs, Nod1 and Nod2 recognize the degradation products of bacterial cell-wall components, and NLRP3 (NALP3) responds to various stimuli to form the inflammasome complex, which promotes the release of IL-1β and IL-18 via caspase-1. Furthermore, cells respond to pathogen-derived dsDNA and incompletely digested self dsDNA by triggering the induction of type I interferon through unidentified pathways, although STING and TBK1 are essential components in dsDNA-triggering signaling10,11,88. DAI (ZBP1-DLM1) has been identified as a putative cytosolic sensor for dsDNA that augments the production of type I interferon in response to dsDNA89. However, the induction of type I interferon by stimulation with dsDNA is intact in DAI-deficient mice, which suggests redundancy90. AIM2, which contains a pyrin and HIN-200 DNA-binding domain, binds dsDNA and forms an inflammasome with ASC to trigger IL-1β production91. AIM2 is required for caspase-1-dependent IL-1β production but is dispensable for the induction of type I interferon in response to dsDNA stimulation, infection with vaccinia virus (a dsDNA virus) and the facultative intracellular Gram-negative bacteria Francisella tularensis that delivers DNA to the host cytoplasm92,93. Moreover, AIM2-deficient mice are more susceptible to lethal infection with F. tularensis than are control mice, probably because of their defects in IL-1β production. Therefore, AIM2 is a crucial component involved in dsDNA-induced production of IL-1β rather than type I interferon. The phosphatase Eya was identified as a molecule that interacts with STING and IPS-1 and enhances the promoter of the gene encoding interferon-β during dsDNA and RLR signaling, although the physiological function of Eya protein remains unclear94. These classes of cytosolic PRRs are expressed by many cells, including immune and non-immune cells such as fibroblasts and epithelial cells, and recognize PAMPs shared with TLRs. Both the specific and overlapping roles of these PRRs have been studied in the context of adaptive immune responses during infection.
Among PRRs, TLRs are expressed mainly on antigen-presenting cells such as DCs and macrophages, as well as on B cells, and many TLR agonists trigger both antibody responses and TH1 and TH17 responses. Several lines of evidence indicate the essential role of TLRs in shaping adaptive immunity. An in vivo model of infection with influenza A virus, which is sensed by TLR7 in pDCs and by RIG-I in other cells, shows that TLR7, rather than RIG-I, is required for mounting B cell and CD4+ T cell responses95. TLR7 has been shown to be critical for protective immunity in a vaccine model. Moreover, TLR7, rather than RIG-I, is required for the differentiation of CD8+ T cells after infection with lymphocytic choriomeningitis virus96. These data collectively suggest that TLR7 contributes to the induction of effective antiviral adaptive immune responses. However, TLRs are insufficient for the robust induction of adaptive immunity, because CD8+ T cell responses to influenza A viruses do not require TLR7 or RLRs. Influenza A viruses are able to activate the NLRP3 inflammasome to trigger IL-1β production, and this pathway is required for the shaping of adaptive immunity97,98,99. Although several mechanisms have been proposed to trigger activation of the NLRP3 inflammasome8,9, it has been shown that influenza A virus–induced activation of the NLRP3 inflammasome is unique in that the virus-encoded M2 ion channel is responsible for triggering this activation100.
Although the relative contributions of cytosolic PRRs to the shaping of adaptive immunity remain unclear, it has been suggested that cytosolic signaling by PRRs in non–antigen presenting cells (non-APCs) is involved in facilitating DC-mediated adaptive immune responses. Poly(I:C) is used as a vaccine adjuvant and is recognized by TLR3 and Mda5. There is high expression of TLR3 by CD8α+ DCs, which have high phagocytic activity for apoptotic virus-infected cells. TLR3-deficient DCs fail to mount CD8+ T cell responses when they engulf either dsRNA-loaded cells or virus-infected cells101. Thus, TLR3 recognition of dsRNA delivered from virus-infected cells in DCs triggers DC maturation and the presentation of viral antigen on major histocompatibility complex class I molecules, which stimulates CD8+ T cell responses (Fig. 2). This process, referred to as 'cross-priming', can be facilitated by type I interferon and other cytokines released by initially infected cells in which Mda5 is involved in virus recognition and cytokine production. Indeed, a study evaluating the adjuvant effects of poly(I:C) in mice deficient in TLR3 and/or Mda5 signaling components has shown that antigen-specific antibody production and the differentiation of CD4+ and CD8+ T cells is regulated via both TLR3- and Mda5-mediated pathways102. Furthermore, a study has shown that recognition of poly(I:C) by Mda5 in DCs, monocytes and stromal cells, with consequent induction of type I interferon, is required for TH1 responses in a mouse model for a human immunodeficiency virus Gag protein vaccine103. This suggests that PRR signaling in both APCs and non-APCs, and the interaction between them, are sufficient for mounting a robust adaptive immune response. The critical role of non-APCs in the establishment of adaptive immunity has also been demonstrated by the study of DNA vaccines that incorporate plasmids with antigenic sequences and other elements that potentiate innate immune responses. The optimal induction of B cell and T cell responses does not require TLR9, RLR or DAI but does require TBK1 and STING88,90. Notably, cell-transfer experiments have demonstrated that responses to dsDNA in both hematopoietic and nonhematopoietic cells are required for the adjuvanticity of DNA vaccines in vivo90.
Endogenous ligands for cell surface TLRs
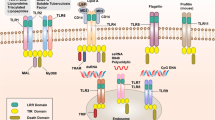
It is becoming increasingly evident that, in addition to responding to PAMPs, TLRs respond to endogenous host molecules and trigger inflammatory responses. Most of these are produced as a result of cell death and injury or by tumor cells, and they include degradation products of the extracellular matrix (ECM), heat-shock proteins and high-mobility group box 1 (HMGB1) proteins, which act as stimulators for cell surface TLRs (Fig. 4). Furthermore, chromatin-DNA and ribonucleoprotein complexes released by dying cells and immune complex–containing self antigens, all of which contain self nucleotides, can stimulate intracellular TLR7 and TLR9 and lead to the development of systemic autoimmune disease (Fig. 4).

Endogenous molecules released by dying cells, such as HMGB1, heat-shock proteins (Hsp) and ECM components, are recognized by TLR2, TLR4 or TLR2-TLR4. Amyloid-β and oxidized LDL (Ox-LDL) are both sensed by TLR4-TLR6 along with the coreceptor CD36. Oxidized (Ox-) phospholipids generated after infection and the antimicrobial peptide β-defensin 2 are recognized by TLR4. Recognition of these endogenous molecules by cell surface TLRs leads to inflammation as well as repair responses. Self DNA and RNA in complex with LL37 are internalized into early endosomes and are recognized by TLR9 and TLR7, respectively. The HMGB1–self DNA complex is internalized via RAGE and is recognized by TLR9. Immune complexes containing self nucleic acids are internalized via Fc receptors, such as FcγRIIa, and stimulate TLR7 and TLR9. Self DNA incompletely digested during apoptosis is probably sensed by an intracellular DNA sensor that activates TBK1. The recognition of self nucleic acids by TLR7, TLR9 and an as-yet-undefined DNA sensor leads to the induction of type I interferon and promotes autoimmune and/or inflammatory diseases.
As a result of injury or inflammation, ECM components are cleaved by cellular proteases and are released outside cells. Some of the released components reportedly activate TLR2 or TLR4 or both (Fig. 4). These include biglycan104, hyaluronic acid105, versican106, extradomain A of fibronectin107 and surfactant protein A108. Biglycan is known to induce the production of inflammatory cytokines and chemokines, and this induction is totally abolished by deficiency in both TLR2 and TLR4. Biglycan-deficient mice are very resistant to zymosan- and LPS-induced shock and are associated with lower TNF concentrations and the infiltration of mononuclear cells into the lungs104, which suggests a role for the biglycan-TLR2-TLR4 pathways in the enhancement of bacteria-induced lung injury. Hyaluronic acid fragments, which accumulate and are released after lung injury, can stimulate macrophages to produce chemokines through TLR2 and TLR4. In a mouse model of noninfectious lung injury in which hyaluronic acid participates, mice doubly deficient in TLR2 and TLR4 show lower survival associated with less recruitment of inflammatory cells, enhanced epithelial cell apoptosis and more tissue injury, which suggests that TLR2-TLR4-mediated recognition of hyaluronic acid promotes inflammation and repair responses105. Extradomain A and surfactant protein A may also be recognized by TLR4 (refs. 107,108).
In addition to ECM components, other cellular components such as HMGB1 and heat-shock proteins serve as ligands for TLRs (Fig. 4). HMGB1, a nuclear non-histone protein that is released by necrotic cells or during inflammation, is a proinflammatory mediator in septic shock and ischemic reperfusion models and is recognized by TLR2, TLR4 and/or TLR9 (ref. 109). Neutralizing antibodies to HMGB1 inhibit damage in a hepatic ischemic reperfusion model, and TLR4-deficient mice show less damage in this model110. That finding suggests that TLR4 responds to endogenous molecules and mediates inflammatory responses in a noninfectious situation. The role of the cytoplasmic HMGB proteins HMGB1, HMGB2 and HMGB3 as universal sentinels for nucleic acids that activate TLRs, RLRs and DNA sensors has been demonstrated111. Heat-shock proteins, including Hsp60, Hsp70, Hsp22 and gp96, have also been linked to the activation of macrophages and DC to induce proinflammatory mediators through TLR2, TLR4 or TLR2-TLR4, although it is still unclear whether or not this effect is due to the contamination of these recombinant protein preparations by Escherichia coli products112.
TLRs also participate in inflammatory responses in certain pathological conditions. In noninfectious inflammatory diseases such as atherosclerosis and Alzheimer's disease, oxidized low-density lipoprotein and amyloid-β, respectively, trigger sterile inflammation and are recognized in a manner dependent on TLR4 and TLR6 (ref. 113; Fig. 4). This recognition is probably achieved via a TLR4-TLR6 heterodimer in cooperation with the scavenger receptor CD36. Versican, an ECM proteoglycan that accumulates in tumor cells, stimulates tumor-infiltrating myeloid cells (via TLR2, TLR6 and CD14) to produce TNF, which accelerates tumor-cell metastasis106. There is a direct interaction between TLR2 and versican. Thus, recognition of versican by TLR2 and the consequent inflammatory environment can support tumor-cell survival.
Infection is also a trigger for the release of endogenous molecules that are recognized by TLRs (Fig. 4). The antimicrobial peptide β-defensin 2, which directly neutralizes invading microbes and is produced in response to infection in mucosal tissue and skin, activates immature DCs via TLR4 to induce the upregulation of costimulatory molecules, which leads to the induction of effective adaptive immune responses114. TLR4- and TRIF-deficient mice are protected from the acute lung injury caused by the administration of inactivated H5N1 avian influenza virus115. This virus triggers production of the oxidized phospholipids that are responsible for acute lung injury through the TLR4-TRIF axis. Thus, the response to oxidative stress via the TLR4 pathway is the key to controlling acute lung injury.
Inappropriate TLR-mediated recognition of self nucleic acids
Self-derived nucleic acids do not activate innate immune responses under normal conditions. Self nucleic acids are properly degraded by serum nucleases before being recognized by TLRs in the endolysosomes. The intracellular localization of TLR7 and TLR9 is important for avoiding contact with extracellular self nucleic acids116. In addition, proteolytic maturation of TLR9 (and possibly TLR7) is important for preventing inappropriate recognition of self DNA by TLR9 leaking to the cell surface. It is possible these safeguards are destroyed in inflammatory and autoimmune conditions. For example, when self-derived nucleic acids form complexes with endogenous proteins, they might become resistant to nucleases and gain access to the endosomal TLRs, which in turn promote and maintain autoimmune processes. Furthermore, the finding that trafficking of TLR7 and TLR9 from the ER to endolysosomes is induced by LPS in addition to their ligands suggests the possibility that inflammation could actually increase the accessibility of nucleic acids to TLR7 and TLR9 (ref. 32).
In systemic lupus erythematosus, there is a higher concentration of antibodies to self nucleic acids or nucleoproteins. Serum from patients with systemic lupus erythematosus promotes pDCs to produce type I interferon, and the concentration of type I interferon correlates with disease severity117. Self nucleic acids or nucleoproteins bound to autoantibodies are internalized via FcγRIIa receptors on DCs and are delivered to TLR7- and TLR9-containing vesicles, which leads to the production of type I interferon118,119 (Fig. 4). Moreover, these immune complexes bind to B cell antigen receptors and are internalized for the activation of these TLRs120, which contributes to the activation of autoreactive B cells. Cooperative activation of DCs and B cells is, therefore, important for the progression and perpetuation of autoimmune diseases.
HMGB1 can bind to both pathogen and self DNA. These HMGB1-DNA complexes bind to the receptor RAGE and are delivered into early endosomes for TLR9 recognition, which results in the activation of pDCs and B cells121 (Fig. 4). LL37 (cathelicidin) is an antimicrobial peptide produced by neutrophils and keratinocytes, and skin lesions of psoriasis have high LL37 expression. LL37 forms aggregates with self DNA and RNA released by necrotic cells, and these aggregates are endocytosed and retained in the early endosomes of pDCs, where they activate TLR9 and TLR7, respectively122,123. Duplication of Tlr7 is found in mice that are hyper-reactive to TLR7 ligands124,125. These mice develop nephritis along with the production of antibodies to RNA-containing autoantigens. Tetramethylpentadecane (pristane) is an isoprenoid alkane found in plants that induces a lupus-like disease in mice when injected into the peritoneal cavity. The ensuing autoantibody production requires interferon signaling. The source of type I interferon production in this case is immature monocytes rather than pDCs, and TLR7, but not RLRs, is indispensable for this126. The U1 small nuclear ribonucleoprotein serves to activate TLR7 in this model, although Fc receptors are dispensable for the production of type I interferon.
Although TLR7 and TLR9 are similar in terms of expression, localization, signaling pathways and target-gene expression, their roles in the pathogenesis of autoimmune disease in mice are distinct127. TLR7 deficiency results in a smaller amount of autoantibodies to RNA-associated antigens in MRL lpr/lpr mice that develop systemic autoimmune diseases such as systemic lupus erythematosus. However, TLR9 deficiency has the opposite effect. This suggests that the TLR9-mediated pathway limits TLR7 responses.
It seems that autoimmune diseases are also caused by inappropriate clearance of self nucleic acids. Mutations in the gene encoding serum DNase I are associated with a lupus-like syndrome in both mice and humans128,129. Mice deficient in lysosomal DNase II accumulate incompletely digested DNA in macrophages, have higher concentrations of type I interferon and TNF and develop autoimmune symptoms such as chronic polyarthritis130. Notably, increases in type I interferon can occur in the absence of DNase II and independently of TLRs. Mutations in the gene encoding the repair exonuclease TREX1 are found in Aicardi-Goutières syndrome, which is characterized by a fatal encephalopathy associated with greater production of type I interferon131,132. Loss of TREX1 in cells results in the accumulation of DNA derived from retro elements, which are responsible for the induction of interferon responses133. Mice with mutations in the gene encoding the endonuclease FEN1 also have more undigested DNA in apoptotic cells and are predisposed to autoimmunity, chronic inflammation and cancer134. Moreover, mutations in the gene encoding SAMHD1, a component of RNase H2, are also associated with Aicardi-Goutières syndrome and have been linked to the negative regulation of antiviral innate immune responses135. Although much less is known about the mechanisms by which these inappropriate nucleic acids cause autoimmunity, cytosolic PRRs for nucleic acids are probably involved (Fig. 4).
Future perspectives
During the past decade, there has been tremendous progress in understanding of the roles of TLRs in pathogen recognition and host defense. Structural analyses of several TLRs have elucidated the mechanisms of PAMP recognition by TLR homo- or heterodimers, and many signaling molecules involved in the activation of NF-κB, AP-1 and IRF proteins have been identified and characterized in detail. The present focus of many investigations is to understand the complex regulation of TLR signaling, including post-transcriptional regulation such as ubiquitination, phosphorylation and mRNA stability, and the spatial and temporal regulation of the TLRs and their signaling complexes. Moreover, the finding that loss of negative regulation is directly linked to inflammatory diseases suggests that pharmacological suppression of TLR responses would be a useful tool for treating these diseases.
The finding that there is activation of TLR signaling during tissue damage in several disease situations in the absence of infection suggests that endogenous molecules serve as TLR agonists, although it is unclear whether this response is biologically important for maintenance of homeostasis, such as tissue repair, or whether this recognition is simply accidental. It is noteworthy that microbial infection triggers the production of modified endogenous molecules (such as HMGB1, oxidized phospholipids, β-defensin 2 and nucleic acids) that are recognized by TLRs or other cytosolic PRRs. This may suggest that these endogenous molecules, along with PAMPs, act as adjuvants to activate innate immune programs via TLRs and/or other PRRs, and have key roles in facilitating adaptive immunity against infecting microbes. Moreover, it has been become apparent that cytosolic PRR signaling in nonimmune cells has a role in promoting or boosting DC maturation and the subsequent activation of antigen-specific T cells. Therefore, the use of PAMPs and/or endogenous agonists that stimulate both immune and non-immune cells will be a helpful tool in the design of effective vaccine adjuvants.
Understanding the complexity of the transcriptional networks that operate during TLR activation and define the subsequent immune response is a major focus of present research. The tools of systems biology are useful here, as this approach has successfully predicted that several transcription factors, such as ATF3 and C/EBPδ, are regulators of IL-6 transcription, and this has been verified in knockout cells56,58. Moreover, comprehensive analyses aimed at understanding the complex TLR transcriptional networks using small interfering RNA have only just begun136. Given that TLR signaling is distinct for individual TLRs, as well as for different cell types, and that microbial pathogens contain multiple PAMPs, the accumulation of knowledge regarding the gene-expression profiles of many cell types after stimulation with each TLR agonist or pathogen will be required. For full understanding of the cooperative roles of the various PRRs in host defense against infection, a combination of in vitro, in vivo and in silico approaches is needed.
References
Akira, S., Takeda, K. & Kaisho, T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2, 675–680 (2001).
Janeway, C.A. Jr. & Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Jin, M.S. & Lee, J.O. Structures of the Toll-like receptor family and its ligand complexes. Immunity 29, 182–191 (2008).
Kawai, T. & Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137 (2006).
Barbalat, R., Lau, L., Locksley, R.M. & Barton, G.M. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10, 1200–1207 (2009).
Yoneyama, M. & Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227, 54–65 (2009).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 (2009).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Ishii, K.J. et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48 (2006).
Stetson, D.B. & Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 (2006).
Akashi-Takamura, S. & Miyake, K. TLR accessory molecules. Curr. Opin. Immunol. 20, 420–425 (2008).
Kim, H.M. et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917 (2007).
Park, B.S. et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 (2009).
Jin, M.S. et al. Crystal structure of the TLR1–TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 (2007).
Kang, J.Y. et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31, 873–884 (2009).
Hoebe, K. et al. CD36 is a sensor of diacylglycerides. Nature 433, 523–527 (2005).
Goodridge, H.S. & Underhill, D.M. Fungal recognition by TLR2 and dectin-1. Handb. Exp. Pharmacol. 183, 87–109 (2008).
Uematsu, S. et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9, 769–776 (2008).
Zhang, D. et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 303, 1522–1526 (2004).
Yarovinsky, F. et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629 (2005).
Choe, J., Kelker, M.S. & Wilson, I.A. Crystal structure of human Toll-like receptor 3 (TLR3) ectodomain. Science 309, 581–585 (2005).
Bell, J.K., Askins, J., Hall, P.R., Davies, D.R. & Segal, D.M. The dsRNA binding site of human Toll-like receptor 3. Proc. Natl. Acad. Sci. USA 103, 8792–8797 (2006).
Kawai, T. & Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci. 1143, 1–20 (2008).
Tabeta, K. et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 101, 3516–3521 (2004).
Zhang, S.Y. et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527 (2007).
Hornung, V. et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 11, 263–270 (2005).
Mancuso, G. et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 10, 587–594 (2009).
Lee, H.K., Lund, J.M., Ramanathan, B., Mizushima, N. & Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401 (2007).
Haas, T. et al. The DNA sugar backbone 2′ deoxyribose determines Toll-like receptor 9 activation. Immunity 28, 315–323 (2008).
Coban, C. et al. Immunogenicity of whole-parasite vaccines against Plasmodium falciparum involves malarial hemozoin and host TLR9. Cell Host Microbe 7, 50–61 (2010).
Kim, Y.M., Brinkmann, M.M., Paquet, M.E. & Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452, 234–238 (2008).
Tabeta, K. et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 7, 156–164 (2006).
Casrouge, A. et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314, 308–312 (2006).
Brinkmann, M.M. et al. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 177, 265–275 (2007).
Kiyokawa, T. et al. A single base mutation in the PRAT4A gene reveals differential interaction of PRAT4A with Toll-like receptors. Int. Immunol. 20, 1407–1415 (2008).
Takahashi, K. et al. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J. Exp. Med. 204, 2963–2976 (2007).
Yang, Y. et al. Heat shock protein gp96 is a master chaperone for Toll-like receptors and is important in the innate function of macrophages. Immunity 26, 215–226 (2007).
Ewald, S.E. et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456, 658–662 (2008).
Park, B. et al. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 9, 1407–1414 (2008).
Asagiri, M. et al. Cathepsin K-dependent Toll-like receptor 9 signaling revealed in experimental arthritis. Science 319, 624–627 (2008).
Matsumoto, F. et al. Cathepsins are required for Toll-like receptor 9 responses. Biochem. Biophys. Res. Commun. 367, 693–699 (2008).
Sepulveda, F.E. et al. Critical role for asparagine endopeptidase in endocytic Toll-like receptor signaling in dendritic cells. Immunity 31, 737–748 (2009).
Peter, M.E., Kubarenko, A.V., Weber, A.N. & Dalpke, A.H. Identification of an N-terminal recognition site in TLR9 that contributes to CpG-DNA-mediated receptor activation. J. Immunol. 182, 7690–7697 (2009).
Kagan, J.C. & Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125, 943–955 (2006).
Rowe, D.C. et al. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc. Natl. Acad. Sci. USA 103, 6299–6304 (2006).
Kagan, J.C. et al. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 (2008).
Tanimura, N., Saitoh, S., Matsumoto, F., Akashi-Takamura, S. & Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 368, 94–99 (2008).
Kawagoe, T. et al. Sequential control of Toll-like receptor–dependent responses by IRAK1 and IRAK2. Nat. Immunol. 9, 684–691 (2008).
Bhoj, V.G. & Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 458, 430–437 (2009).
Yamamoto, M. et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 7, 962–970 (2006).
Tokunaga, F. et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 11, 123–132 (2009).
Rahighi, S. et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136, 1098–1109 (2009).
Lo, Y.C. et al. Structural basis for recognition of diubiquitins by NEMO. Mol. Cell 33, 602–615 (2009).
Yamamoto, M. et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature 430, 218–222 (2004).
Litvak, V. et al. Function of C/EBPδ in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 10, 437–443 (2009).
Kuwata, H. et al. IκBNS inhibits induction of a subset of Toll-like receptor-dependent genes and limits inflammation. Immunity 24, 41–51 (2006).
Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178 (2006).
Pobezinskaya, Y.L. et al. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 9, 1047–1054 (2008).
Ermolaeva, M.A. et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 9, 1037–1046 (2008).
Chang, M., Jin, W. & Sun, S.C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 10, 1089–1095 (2009).
Hacker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 357, re13 (2006).
Hacker, H. et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207 (2006).
Oganesyan, G. et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439, 208–211 (2006).
Tseng, P.H. et al. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 11, 70–75 (2010).
Wang, C. et al. The E3 ubiquitin ligase Nrdp1 'preferentially' promotes TLR-mediated production of type I interferon. Nat. Immunol. 10, 744–752 (2009).
Shinohara, M.L. et al. Osteopontin expression is essential for interferon-α production by plasmacytoid dendritic cells. Nat. Immunol. 7, 498–506 (2006).
Cao, W. et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat. Immunol. 9, 1157–1164 (2008).
Takaoka, A. et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249 (2005).
Tsujimura, H. et al. Toll-like receptor 9 signaling activates NF-κB through IFN regulatory factor-8/IFN consensus sequence binding protein in dendritic cells. J. Immunol. 172, 6820–6827 (2004).
Honda, K. et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434, 1035–1040 (2005).
Palsson-McDermott, E.M. et al. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88–independent TLR4 pathway. Nat. Immunol. 10, 579–586 (2009).
Carty, M. et al. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 7, 1074–1081 (2006).
Shi, M. et al. TRIM30α negatively regulates TLR-mediated NF-κB activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 9, 369–377 (2008).
Tanaka, T., Grusby, M.J. & Kaisho, T. PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 8, 584–591 (2007).
Kayagaki, N. et al. DUBA: a deubiquitinase that regulates type I interferon production. Science 318, 1628–1632 (2007).
Taganov, K.D., Boldin, M.P., Chang, K.J. & Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 103, 12481–12486 (2006).
Sheedy, F.J. et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 11, 141–147 (2010).
Kawagoe, T. et al. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat. Immunol. 10, 965–972 (2009).
Hampe, J. et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211 (2007).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008).
Cadwell, K. et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 (2008).
Matsushita, K. et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458, 1185–1190 (2009).
Carrick, D.M., Lai, W.S. & Blackshear, P.J. The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis Res. Ther. 6, 248–264 (2004).
Turer, E.E. et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J. Exp. Med. 205, 451–464 (2008).
Croker, B.A. et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc. Natl. Acad. Sci. USA 105, 15028–15033 (2008).
An, H. et al. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat. Immunol. 9, 542–550 (2008).
Ishikawa, H., Ma, Z. & Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Takaoka, A. et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007).
Ishii, K.J. et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 (2008).
Hornung, V. & Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 10, 123–130 (2010).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. advance online publication, doi:10.1038/ni1859 (28 March 2010).
Rathinam, V.A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. advance online publication, doi:10.1038/ni.1864 (29 March 2010).
Okabe, Y., Sano, T. & Nagata, S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature 460, 520–524 (2009).
Koyama, S. et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J. Immunol. 179, 4711–4720 (2007).
Jung, A. et al. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 82, 196–206 (2008).
Allen, I.C. et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565 (2009).
Thomas, P.G. et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575 (2009).
Ichinohe, T., Lee, H.K., Ogura, Y., Flavell, R. & Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87 (2009).
Ichinose, T., Pang, I.K. & Iwasaki, A. Influenza virus activates inflammasomes through intracellular M2 channel. Nat. Immunol. advance online publication, doi:10.1038/ni.1861 (11 April 2010).
Schulz, O. et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433, 887–892 (2005).
Kumar, H., Koyama, S., Ishii, K.J., Kawai, T. & Akira, S. Cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J. Immunol. 180, 683–687 (2008).
Longhi, M.P. et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 206, 1589–1602 (2009).
Schaefer, L. et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Invest. 115, 2223–2233 (2005).
Jiang, D. et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 11, 1173–1179 (2005).
Kim, S. et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106 (2009).
Okamura, Y. et al. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276, 10229–10233 (2001).
Guillot, L. et al. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J. Immunol. 168, 5989–5992 (2002).
Yang, H. & Tracey, K.J. Targeting HMGB1 in inflammation. Biochim. Biophys. Acta 1799, 149–156 (2009).
Tsung, A. et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201, 1135–1143 (2005).
Yanai, H. et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103 (2009).
Tsan, M.F. & Gao, B. Heat shock proteins and immune system. J. Leukoc. Biol. 85, 905–910 (2009).
Stewart, C.R. et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 (2010).
Biragyn, A. et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298, 1025–1029 (2002).
Imai, Y. et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008).
Barton, G.M., Kagan, J.C. & Medzhitov, R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 7, 49–56 (2006).
Marshak-Rothstein, A. & Rifkin, I.R. Immunologically active autoantigens: the role of Toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol. 25, 419–441 (2007).
Means, T.K. et al. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115, 407–417 (2005).
Vollmer, J. et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J. Exp. Med. 202, 1575–1585 (2005).
Viglianti, G.A. et al. Activation of autoreactive B cells by CpG dsDNA. Immunity 19, 837–847 (2003).
Tian, J. et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 (2007).
Lande, R. et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569 (2007).
Ganguly, D. et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 206, 1983–1994 (2009).
Pisitkun, P. et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672 (2006).
Fairhurst, A.M. et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur. J. Immunol. 38, 1971–1978 (2008).
Lee, P.Y. et al. TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. J. Exp. Med. 205, 2995–3006 (2008).
Christensen, S.R. et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428 (2006).
Napirei, M. et al. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 25, 177–181 (2000).
Yasutomo, K. et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28, 313–314 (2001).
Kawane, K. et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 (2006).
Crow, Y.J. et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920 (2006).
Lee-Kirsch, M.A. et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067 (2007).
Stetson, D.B., Ko, J.S., Heidmann, T. & Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008).
Zheng, L. et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 13, 812–819 (2007).
Rice, G.I. et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 41, 829–832 (2009).
Amit, I. et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–263 (2009).
Acknowledgements
We thank E. Kamada for secretarial assistance. Supported by the Special Coordination Funds of the Japanese Ministry of Education, Culture, Sports, Science and Technology, the 21st Century Center of Excellence Program of Japan and the National Institutes of Health (P01 AI070167).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Kawai, T., Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11, 373–384 (2010). https://doi.org/10.1038/ni.1863
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.1863
This article is cited by
-
Understanding immune microenvironment alterations in the brain to improve the diagnosis and treatment of diverse brain diseases
Cell Communication and Signaling (2024)
-
Exploring the genetic factors behind the discrepancy in resistance to bovine tuberculosis between African zebu cattle and European taurine cattle
Scientific Reports (2024)
-
Formation of memory assemblies through the DNA-sensing TLR9 pathway
Nature (2024)
-
A second-generation M1-polarized CAR macrophage with antitumor efficacy
Nature Immunology (2024)
-
Cyclical palmitoylation regulates TLR9 signalling and systemic autoimmunity in mice
Nature Communications (2024)
