
Abstract
Pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs) and RIG-like helicase (RLH) receptors, are involved in innate immune antiviral responses. Here we show that nucleotide-binding oligomerization domain 2 (Nod2) can also function as a cytoplasmic viral PRR by triggering activation of interferon-regulatory factor 3 (IRF3) and production of interferon-β (IFN-β). After recognition of a viral ssRNA genome, Nod2 used the adaptor protein MAVS to activate IRF3. Nod2-deficient mice failed to produce interferon efficiently and showed enhanced susceptibility to virus-induced pathogenesis. Thus, the function of Nod2 as a viral PRR highlights the important function of Nod2 in host antiviral defense mechanisms.
Similar content being viewed by others
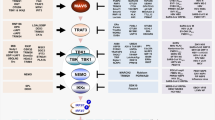


Main
Innate immune antiviral responses are the first line of defense against viral infection1,2. Interferon-α/β (IFN-α/β) serves an important function during innate antiviral responses by activating the kinase Jak–transcription factor STAT signaling pathway3. Virus-infected cells use pattern-recognition receptors (PRRs) to recognize pathogen-associated molecular patterns (PAMPs; in this case, virus-associated molecular patterns) and to trigger phosphorylation of the transcription factor interferon-regulatory factor 3 (IRF3), which then translocates to the nucleus to transactivate interferon genes4. So far two classes of viral PRRs have been identified: the Toll-like receptors (TLRs)5 and the RIG-like helicase (RLH) receptors such as RIG-I and Mda5 (ref. 6).
A third class of PRR includes members of the nucleotide-binding domain (NBD)– and leucine-rich-region (LRR)–containing family of cytoplasmic proteins (known as NLRs); these proteins respond to bacterial PAMPs to activate the transcription factor NF-κB and mitogen-activated protein kinase pathways7,8,9. For example, the NLR Nod2 detects bacterial PAMPs, including muramyl dipeptide (MDP)10. However, so far, no NLRs have been reported to respond to virus-specific PAMPs and activate an antiviral response.
It has been shown that the NLR family member NLRX1 interacts with the mitochondrial antiviral signaling (MAVS) protein (also known as IPS-1, VISA or CARDIF) to negatively regulate the interferon pathway11 and induces the formation of reactive oxygen species12. Thus, members of NLR family of proteins may modulate (either positively or negatively) the host antiviral apparatus. In addition, Nod2 facilitates production of human β-defensin 2 after stimulation with MDP, and human β-defensin 2 is also upregulated in human cells infected with respiratory syncytial virus (RSV), a paramyxovirus that has a negative-sense single-stranded RNA (ssRNA) genome13,14.
Here, to investigate the involvement of NLRs in innate antiviral response, we examined the ability of various NLRs to activate IRF3 and interferon after infection with RSV, which causes severe lung disease in infants and children and elderly and immunocompromised people15,16. Among the various NLRs, Nod2 activated IRF3 and interferon in RSV-infected cells. Activation of Nod2 by ssRNA resulted in signaling dependent on MAVS. Both synthetic ssRNA and viral ssRNA genome were able to activate the MAVS-IRF3-interferon pathway. The important physiological function of Nod2 in antiviral defense was evident from the enhanced RSV pathogenesis, lung disease and viral susceptibility of Nod2-deficient mice. We found similar functions for Nod2 in responses to influenza A and parainfluenza viruses. Thus, our studies demonstrate a previously unknown function for Nod2 as a viral PRR important for the host defense against virus infection.
Note: Supplementary information is available on the Nature Immunology website.
Results
Induction of interferon production by ssRNA via Nod2
To study the involvement of NLR proteins in antiviral responses, we expressed various hemagglutinin (HA)-tagged human NLR proteins (such as Nod1, Nod2, IPAF, NAIP and Nod3) in 293 human embryonic kidney cells, which do not endogenously express most NLRs. We treated these cells with synthetic ssRNA and analyzed interferon production and IRF3 activation. We used ssRNA because several highly pathogenic viruses, including paramyxoviruses (RSV, Sendai virus, human parainfluenza viruses and measles virus), rhabdoviruses (rabies virus and vesicular stomatitis virus) and orthomyxoviruses (influenza viruses) have ssRNA genomes and because ssRNA activates PRRs, including TLR7, TLR8 and RIG-I (refs. 5,6). Nod2 but not Nod1 facilitated ssRNA-induced activation of IRF3 (Fig. 1a) and IFN-β (Supplementary Fig. 1a,b). Such activation was lacking in cells treated with CpG DNA (Fig. 1a and Supplementary Fig. 1a,b). Transfected 293 cells had high expression of HA-tagged Nod constructs (Supplementary Fig. 1c); in addition, HA-Nod2 and HA-Nod1 proteins were functional, as they facilitated activation of NF-κB in 293 cells treated with MDP or γ-D-glutamyl-meso-diaminopimelic acid, respectively (Supplementary Fig. 1d).

(a) Activation of an IRF3 luciferase reporter in 293 cells transfected with pcDNA, human HA-Nod1 or human HA-Nod2 and left untreated (UT) or treated for 6 h with ssRNA or CpG DNA. (b) RT-PCR analysis of Nod2 expression in A549 cells left untreated or stimulated with ssRNA (time, above lanes). (c) Activation of an IFN-β luciferase reporter in A549 cells transfected with control siRNA or Nod2-specific siRNA and left untreated or stimulated for 6 h with ssRNA. (d,e) Enzyme-linked immunosorbent assay (ELISA) of IFN-β production by wild-type (WT) and Nod2-deficient (Nod2-KO) BMMs (d) or MEFs (e) left untreated or stimulated with ssRNA (time, below graphs). Data are representative of three independent experiments (a,c–e; mean ± s.d.) or three experiments (b).
To establish the physiological relevance of Nod2-mediated, ssRNA-induced activation of IRF3, we next evaluated the function of endogenous Nod2 in inducing IRF3-interferon after ssRNA treatment. For these studies we used human lung epithelial A549 cells, as these cells are permissive to most viruses that have ssRNA genomes and endogenously express various PRRs. Treatment of these cells with ssRNA resulted in higher expression of Nod2 (Fig. 1b); Nod1 expression remained unchanged (data not shown). Nod2-specific small interfering RNA (siRNA) diminished Nod2 expression in ssRNA-treated A549 cells (Supplementary Fig. 2a) and impaired ssRNA-induced activation of IRF3 and IFN-β (Fig. 1c and Supplementary Fig. 2b).
Similarly, ssRNA-induced production of IFN-β was lower in bone marrow–derived macrophages (BMMs) and mouse embryo fibroblasts (MEFs) isolated from Nod2-deficient than in those from wild-type mice (Fig. 1d,e). In contrast, wild-type and Nod2-deficient BMMs and MEFs produced similar amounts of IFN-β after treatment with poly(I:C) (double-stranded RNA that activates TLR3; Supplementary Fig. 2c). The ability of poly(I:C) to induce interferon (via TLR3) in both wild-type and Nod2-deficient MEFs showed that the IRF3-interferon pathway is intact in Nod2-deficient mice. Furthermore, administration of poly(I:C) in vivo resulted in the production of similar concentrations of IFN-β in wild-type and Nod2-deficient mice (Supplementary Fig. 2d). Thus, our results obtained with cell lines and primary cells demonstrate that activation of Nod2 by ssRNA results in IFN-β production.
Nod2 facilitates virus-induced interferon production
To further confirm the ability of Nod2 to launch an antiviral response, we infected 293 cells with RSV. RSV induced activation of IRF3 and IFN-β in 293 cells expressing HA-Nod2 but not in 293 cells expressing HA-Nod1 (Fig. 2a,b). Inactivation of virion particles with ultraviolet light abolished the ability of RSV to activate IRF3 in Nod2-expressing cells (Fig. 2a). The inability of RSV inactivated by ultraviolet light to activate IRF3 indicated that an intact viral RNA genome is essential for Nod2 activation. The direct function of viral components in Nod2 activation was further confirmed by the loss of IRF3 activation after inhibition of RSV cellular entry with an RSV-neutralizing antibody (specific for the RSV fusion protein; Supplementary Fig. 2e).

(a,b) Activation of IRF3 (a) and IFN-β (b) luciferase reporter genes in mock-infected and RSV-infected 293 cells expressing pcDNA, HA-Nod1 or HA-Nod2. (a) Luciferase activity 6 h after infection in cells with (+UV) or without (−UV) treatment with ultraviolet irradiation. (b) Time after infection, below graph. (c) Plaque assay of VSV infectivity in 293 cells expressing pcDNA, HA-Nod1 or HA-Nod2, presented as crystal violet staining (left) and VSV titer (right; plaque-forming units (PFU) per ml). (d) RSV infectivity in 293 cells expressing pcDNA, HA-Nod1 or HA-Nod2, presented relative to viral titer in cells expressing pcDNA, set as 100%. Data are representative of three independent experiments (mean ± s.d.).
We established the functional importance of Nod2 in antiviral responses with the interferon-sensitive vesicular stomatitis virus (VSV). Plaque assay of VSV titers obtained from 293 cells expressing HA-Nod2 or HA-Nod1 showed that cells expressing HA-Nod2 had much lower viral titers (Fig. 2c). Like VSV, RSV titers were lower in Nod2-expressing cells than in Nod1-expressing cells (Fig. 2d). Human parainfluenza virus type 3 (ref. 17) and VSV18 also activated IRF3 in Nod2-expressing 293 cells (Supplementary Fig. 3a). In contrast, vaccinia virus, a DNA virus, failed to activate IRF3 in Nod2-expressing cells (Supplementary Fig. 3b). These results demonstrated that like the RLH receptors RIG-I and Mda5, Nod2 can function as a cytoplasmic PRR for a viral ssRNA genome (viral ssRNA).
We next evaluated the function of endogenous Nod2 in inducing IRF3-interferon in response to RSV infection. Published studies have described IRF3 activation and IFN-β production in RSV-infected A549 cells as early as 2 h after infection19,20. Although uninfected A549 cells did not have detectable expression of Nod2, RSV infection resulted in higher Nod2 expression within 2 h of infection (Fig. 3a). RSV infection failed to induce Nod1 expression (data not shown). Nod2-specific siRNA markedly diminished Nod2 expression after RSV infection (Supplementary Fig. 4a) and resulted in less activation of IRF3 and IFN-β after RSV infection (Fig. 3b and Supplementary Fig. 4b). The effect was more pronounced during early time points after infection (4 h and 6 h) than later (10 h) after infection (Fig. 3b). This result suggests that Nod2 is critical for the early antiviral response, whereas during late time periods after infection, other PRRs (such as RIG-I) may activate the IRF3-interferon pathway.

(a) RT-PCR analysis of Nod2 expression in mock- and RSV-infected A549 cells. (b) Activation of an IRF3 luciferase reporter in mock- and RSV-infected A549 cells transfected with control or Nod2-specific siRNA (time after infection, below graph). (c) ELISA of IFN-β production by mock- and RSV-infected primary NHBE cells transfected with control or Nod2-specific siRNA. (d–f) ELISA of IFN-β production by mock- and RSV-infected alveolar macrophages (d), BMMs (e) and MEFs (f) isolated from wild-type or Nod2-deficient mice. Data are representative of three experiments (a) or three independent experiments (b–f; mean ± s.d.).
Indeed, as shown before20, we found that RIG-I expression in A549 cells was detectable only at late time points after infection with RSV (Supplementary Fig. 4c). In addition, the early antiviral response was independent of RIG-I, because silencing of RIG-I had no effect in IFN-β expression during early RSV infection (Supplementary Fig. 4d,e). Like A549 cells, MEFs did not express abundant Nod2 until early time points after RSV infection, and RIG-I was undetectable until late time points after RSV infection (Supplementary Fig. 4f,g). Thus, temporal expression of Nod2 and RIG-I during early and late infection, respectively, may facilitate optimal sustained interferon production by virus-infected cells.
Next we examined the function of Nod2 in primary normal human bronchial epithelial cells (NHBE cells), as these cells constitute the main cell type infected by RSV in humans. RSV rapidly induced Nod2 expression in NHBE cells (Supplementary Fig. 5). Nod2 was essential for interferon production, as Nod2-specific siRNA markedly diminished IFN-β production by RSV-infected NHBE cells (Fig. 3c). We further established the critical function of Nod2 by demonstrating that BMMs, MEFs and alveolar macrophages derived from Nod2-deficient mice produced less IFN-β than did their wild-type counterparts after RSV infection (Fig. 3d–f). Nod2-deficient MEFs and BMMs also showed defective IFN-β production after infection with influenza A/PR/8/34 (H1N1) virus (Supplementary Fig. 6). Collectively these data show an important function for endogenous Nod2 in the induction of antiviral immune responses.
Nod2 interacts with viral ssRNA
We next investigated the function of viral ssRNA in Nod2 activation. Viral ssRNA genome isolated from purified RSV virion particles activated IRF3 only in Nod2-expressing cells (Fig. 4a). An intact ssRNA genome was required for Nod2 activation, as treatment of the viral ssRNA with RNAse abolished IRF3 activation (Fig. 4a). Viral ssRNA uses Nod2 for interferon production, as we found less IFN-β production after viral ssRNA treatment in MEFs and BMMs isolated from Nod2-deficient mice than in those from wild-type mice (Fig. 4b,c).

(a) Activation of an IRF3 luciferase reporter in 293 cells expressing pcDNA, HA-Nod1 and HA-Nod2 that were left untreated or stimulated with RSV viral ssRNA; + RNAse (below graph), viral ssRNA treated with RNAse. (b,c) ELISA of IFN-β production by wild-type or Nod2-deficient BMMs (b) and MEFs (c) left untreated or stimulated with viral ssRNA. (d) Association of Nod2 with RNA in 293 cells transfected with pcDNA or HA-Nod2 and then mock infected or infected with RSV; at 4 h or 8 h after infection, Nod2 was immunoprecipitated with anti-HA agarose and bound RNA was amplified with primers specific for RSV nucleocapsid protein (top) or GAPDH (bottom), followed by agarose gel electrophoresis. (e) Cell-free assay of HA-Nod2 bound to anti-HA agarose beads, incubated with RSV ssRNA (left) or total cellular mRNA (right); bound RNA was amplified with the primers in d, followed by agarose gel electrophoresis. Far right, total cellular mRNA amplified with GAPDH-specific primer (positive control). Data are representative of three independent experiments (a–c; mean ± s.d.) or two experiments (d,e).
The function of viral ssRNA as an activator of Nod2 was further demonstrated by the interaction of viral ssRNA with Nod2 in a cellular milieu. We immunoprecipitated HA-Nod2 from RSV-infected 293 cells transfected with HA-Nod2 and amplified bound RNA with primers specific for either glyceraldehyde phosphate dehydrogenase (GAPDH; control) or RSV nucleocapsid protein. These experiments demonstrated association of Nod2 with viral RNA but not with control RNA (Fig. 4d). In a cell-free assay, we incubated HA-Nod2 bound to HA-agarose beads with RSV ssRNA genome or mRNA isolated from cells. After incubation, we amplified bound RNA with primers specific for either GAPDH or RSV nucleocapsid protein; we detected interaction of Nod2 with RSV ssRNA (Fig. 4e). In contrast, GAPDH mRNA (total cellular mRNA is enriched for this) did not associate with Nod2 (Fig. 4e). We also noted a failure of Nod1 to interact with viral ssRNA (data not shown). These results demonstrate that interaction of viral ssRNA with Nod2 results in its activation and subsequent induction of interferon production.
MAVS is required for Nod2-mediated responses
We next focused on the mechanism used by Nod2 to activate IRF3-interferon. As both RIG-I and Nod2 have caspase-recruitment domains (CARDs)6,7,8,9, we speculated that similar to RIG-I, Nod2 may also interact with MAVS. In addition, a study has shown that the NLR family member NLRX1 interacts with mitochondria-localized MAVS through its NBD, a domain also found in Nod2 (ref. 11). The interaction of Nod2 with MAVS was essential for Nod2-mediated activation of antiviral responses, as MAVS-specific siRNA (Supplementary Fig. 7) and Nod2-specific siRNA (Fig. 5a) diminished RSV-induced production of IFN-β from infected NHBE cells to a similar extent. We obtained similar results with MEFs derived from MAVS-deficient mice infected with RSV or transfected with viral or synthetic ssRNA (Fig. 5b,c). Influenza A virus also required MAVS for interferon production, as IFN-β production by Nod2-deficient and MAVS-deficient MEFs was diminished to a similar extent after influenza A infection (Supplementary Fig. 8). These results demonstrate that MAVS is critical for virus-induced Nod2-mediated interferon production.

(a) ELISA of IFN-β production by mock- and RSV-infected primary NHBE cells transfected with control, Nod2-specific or MAVS-specific siRNA. (b) ELISA of IFN-β production by mock- and RSV-infected MEFs isolated from wild-type, Nod2-deficient or MAVS-deficient (MAVS-KO) mice. (c) ELISA of IFN-β production by MEFs left untreated or stimulated with synthetic ssRNA or viral (RSV) ssRNA. Data are representative of three independent experiments (mean ± s.d.).
We next examined interaction of Nod2 with MAVS. Initially we investigated the ability of activated Nod2 to translocate to the mitochondria. Immunoblot analysis of mitochondrial extracts from RSV-infected Nod2-expressing cells showed that although approximately 6–7% of Nod2 was located in mitochondria in uninfected cells, RSV infection resulted in enrichment of Nod2 in mitochondria (40–45% of total cellular Nod2; Supplementary Fig. 9a,b). Immunofluorescence analysis also showed localization of endogenous Nod2 together with mitochondria in RSV-infected A549 cells (Supplementary Fig. 9c). To study the interaction of Nod2 with MAVS, we transfected 293 cells with HA-Nod2 and green fluorescent protein (GFP)-tagged MAVS. Coimmunoprecipitation analysis showed interaction of Nod2 with MAVS (Fig. 6a), and this interaction was enhanced after RSV infection. Cells had high expression of GFP-MAVS, as detected by immunoblot analysis of cell lysates with antibody to GFP (anti-GFP; Fig. 6a), and a substantial amount of HA-Nod2 was bound to the anti-HA–agarose beads used to precipitate the HA-Nod2–GFP-MAVS complex (Fig. 6b). In contrast to Nod2, Nod1 failed to interact with MAVS (Supplementary Fig. 9d). Double-labeling immunofluorescence studies with RSV-infected 293 cells expressing GFP-MAVS and HA-Nod2 confirmed the colocalization of Nod2 and MAVS (Fig. 6c). Similarly, RSV infection of A549 cells (Fig. 6d) and NHBE cells (Fig. 6e) enhanced the colocalization of endogenous Nod2 with endogenous MAVS. These results demonstrate that Nod2 interacts with MAVS during virus infection.

(a) Immunoblot (IB) analysis of proteins immunoprecipitated (IP) with anti-HA–agarose beads from lysates of 293 cells transfected with pcDNA, HA-Nod2 and/or GFP-MAVS and mock infected or infected with RSV, probed with anti-GFP. Bottom, immunoblot analysis of 25 μg total cellular lysate with anti-GFP. (b) Immunoblot analysis of GFP-MAVS and HA-Nod2 in lysates (25 μg total cellular lysate protein probed with anti-GFP and anti-HA; top) and of HA-Nod2 bound to anti-HA agarose beads (bottom). (c) Confocal microscopy of RSV-infected (4 h) 293 cells coexpressing GFP-MAVS (green) and HA-Nod2 (red). (d,e) Confocal microscopy of mock- or RSV-infected (6 h) A549 cells (d) or RSV-infected (4 h) NHBE cells (e) stained with anti-Nod2 and anti-MAVS. Original magnification, ×40 (c, top row), ×20 (c, bottom row), ×40 (d), ×10 (e, top and middle rows) or ×40 (e, bottom row). Data are representative of three (a,b) or two (c–e) experiments.
Nod2 can activate the NF-κB and mitogen-activated protein kinase pathways through the kinase RICK (also known as Rip2, CARDIAK, CCK or Ripk2)21,22. Bacterial products such as MDP specifically stimulate Nod2 and result in NF-κB activation via RICK. However, treatment of Nod2-expressing 293 cells with MDP did not activate IRF3 (Supplementary Fig. 10a). Likewise, treatment of NHBE cells and BMMs with MDP did not result in IFN-β production (Supplementary Fig. 10b,c). In addition, RICK may not be important in interferon induction by Nod2, as silencing of endogenous RICK expression did not alter Nod2-mediated production of IFN-β in RSV-infected cells (Supplementary Fig. 11). We also examined the efficiency of the interaction between Nod2 and MAVS relative to that of RIG-I and MAVS. Although both Nod2 and RIG-I associated with MAVS, the RIG-I–MAVS interaction was slightly more efficient than the Nod2-MAVS interaction (Supplementary Fig. 12a). In addition, we also noted that in RSV-infected cells, Nod2 interacted more efficiently with MAVS than with RICK (Supplementary Fig. 12b). However, Nod2 efficiently interacted with RICK in cells stimulated with the well-established Nod2 stimulator MDP (Supplementary Fig. 12b). Thus, we speculate that Nod2 uses either RICK or MAVS, depending on the stimulus (for example, MDP versus ssRNA) to activate either IRF3 or NF-κB.
In addition to IRF3, NF-κB activation is required for the expression of interferon genes. Although IRF3 alone is able to induce the transcription of interferon genes, the transactivating function of NF-κB acts synergistically with IRF3 to promote optimal interferon expression23. This is also true for Nod2-mediated interferon expression, as suppressing NF-κB activity in RSV-infected cells diminished interferon expression via activated Nod2 by 30–35%; as expected, expression of the NF-κB-dependent TNF gene in RSV-infected cells was diminished by 80% (Supplementary Fig. 13). On the basis of these results, we speculate that Nod2 activated by stimulation with viral ssRNA interacts with MAVS to induce activation of both IRF3 and NF-κB in a way similar to that of RLHs6. In contrast, Nod2 activated by bacterial products (such as MDP) activates NF-κB via RICK. Further detailed studies are needed to investigate the function of Nod2 in activating the NF-κB pathway during virus infection and the function of MAVS and RICK during these events.
MAVS interacts with the LRR-NBD domains of Nod2
The CARD of RIG-I promotes its association with MAVS, whereas NLRX1 uses its NBD to interact with MAVS. Thus, we next investigated the function of the NBD, CARD and LRR domains of Nod2 in MAVS association. For these studies, we generated various Nod2 deletion mutants tagged with both histidine (His) and Myc:ΔCARD (a Nod2 mutant lacking both CARDs), ΔNBD (a Nod2 mutant lacking the NBD) and ΔLRR (a Nod2 mutant lacking the LRR domain; Fig. 7a). We expressed these mutants in 293 cells along with GFP-MAVS. We obtained lysates of these cells and precipitated the lysates with nickel-agarose beads and, after washing the beads, analyzed the proteins bound to the beads by immunoblot with anti-GFP. Although ΔCARD was able to interact with MAVS, neither ΔNBD nor ΔLRR associated with MAVS (Fig. 7b). The various Nod2 mutant–expressing cells expressed similar amounts of GFP-MAVS (Fig. 7c). In addition, similar amounts of His-Myc–tagged Nod2 mutants were bound to the nickel-agarose beads in the experimental conditions used to study the interaction of MAVS with the Nod2 mutants (Fig. 7d). These results indicate that unlike RIG-I, the CARDs of Nod2 are not important for its interaction with MAVS. However, the NBD and LRR domains of Nod2 are required for the MAVS association. We further confirmed that conclusion by examining the functionality of the Nod2 mutants in activating IRF3. Infection of 293 cells transfected with either wild-type or mutant (ΔCARD, ΔNBD or ΔLRR) Nod2 constructs showed that wild-type Nod2 and ΔCARD induced IRF3 activation after infection with RSV, but neither ΔNBD nor ΔLRR did so (Supplementary Fig. 14).

(a) Nod2 constructs with deletion of specific domains. (b) Binding of Nod2 to MAVS in 293 cells expressing various His-Myc–tagged Nod2 constructs or pcDNA along with GFP-MAVS; cell lysates were incubated with nickel-agarose beads (Ni-agarose), then beads were washed and bound proteins were analyzed by immunoblot with anti-GFP. P, precipitation. (c) Immunoblot analysis of the expression of GFP-MAVS lysates of the cells in b, probed with anti-GFP. Far left, cells transfected with pcDNA only. (d) Immunoblot analysis of lysates of 293 cells expressing wild-type Nod2 or His-Myc–tagged Nod2 constructs along with GFP-MAVS, incubated with nickel-agarose, probed with anti-Myc to detect Nod2 constructs bound to the nickel-agarose beads. Arrowheads indicate wild-type Nod2 and deletion constructs of Nod2. Data are representative of three experiments.
Function of Nod2 in the host antiviral defense
Finally, we assessed the physiological function of Nod2 by infecting wild-type and Nod2-deficient mice with RSV. The mouse model of RSV infection mimics virus infection in humans, as infected mice can develop disease states resembling pneumonia24,25; in addition, RSV-infected mice induce a robust antiviral response in the respiratory tract characterized by production of IFN-β and expression of interferon-dependent genes such as Mx1 during early RSV infection (within 12 h of infection)24,25,26,27,28,29,30. Moreover, RSV is sensitive to interferon in infected mice, as a dose of interferon as low as 200 units per ml inhibits RSV infection in mice by 100-fold (ref. 26). During RSV infection of the mouse respiratory tract, interferon is induced early during infection (at 12 h to 2 d after infection), but its production is lost at 3 d after infection24,25,26,27,28,29,30. This observation suggests that interferon is important in restricting the spread of RSV during early infection and that the production of interferon dictates the clinical outcome of the disease (for example, lung inflammation and apoptosis of airway cells).
We infected wild-type and Nod2-deficient mice with a sublethal dose of RSV (5 × 106 plaque-forming units per mouse, delivered by intranasal inoculation), then collected lungs and bronchoalveolar lavage (BAL) fluid at various times. We found expression of mouse Nod2 in RSV-infected lungs at 1 d after infection, and this expression was lost at 4 d after infection (Supplementary Fig. 15a). This result suggests an important function for Nod2 in interferon expression, as the interferon-induction kinetics correlated with the Nod2-expression kinetics24,25,26,27,28,29,30. Nod2-deficient mice had lower IFN-β production in the respiratory tract and higher viral titers than did wild-type mice (Fig. 8a,b).

(a) IFN-β concentrations in BAL fluid of mock- or RSV-infected wild-type and Nod2-deficient mice (n = 4 mice per group). P < 0.05 (t-test for data normally distributed; Mann-Whitney rank sum test for data not normally distributed). (b) RSV titers in the BAL fluid of wild-type and Nod2-deficient mice at 3 d after infection. P < 0.05 (t-test for data normally distributed; Mann-Whitney rank sum test for data not normally distributed). (c) Hematoxylin and eosin staining of lung sections from mock- or RSV-infected wild-type and Nod2-deficient mice. Original magnification, ×10. (d) Neutrophil sequestration in lungs, assessed by MPO activity assay of total lung homogenates of RSV-infected wild-type and Nod2-deficient mice (n = 5 mice per group; 10 mice total) at 2 d after infection, presented relative to activity in mock-infected mice. P < 0.05. (e) TUNEL staining of lung sections from RSV-infected wild-type and Nod2-deficient mice. Original magnification, ×10. (f) Survival of wild-type and Nod2-deficient mice infected with RSV (5 × 108 PFU per mouse). P > 0.02, Nod2-deficient versus wild-type (Wilcoxon test). Data are representative of three (a,b,d) or four (c,e,f) experiments (mean ± s.e.m in a,b,d).
It is well known that RSV causes lung disease by inducing pneumonia, a massive inflammation of the lungs31. A larger viral burden ultimately results in enhanced inflammation and exaggerated lung disease due to flooding of alveolar spaces with edema fluid. This occurs as a result of enhanced permeability of the epithelial barrier due to apoptosis of airway epithelial cells. RSV infection resulted in more severe lung pathology in Nod2-deficient mice, as shown by staining of lung sections with hematoxylin and eosin at 3 and 5 d after infection (Fig. 8c). We noted massive peribronchial lymphocytic inflammation and filling of the lumen with exudates of infiltrating neutrophils and mucus. Neutrophils constitute the main immune cells infiltrating the lung of RSV-infected mice and humans and a large number of these cells in the airway causes severe immunopathology associated with RSV clinical disease32,33,34. To examine neutrophil accumulation in the lungs, we did a myeloperoxidase (MPO) activity assay35,36 with lung homogenates of RSV-infected wild-type and Nod2-deficient mice. Greater RSV-induced enhancement of neutrophil activity was visible in the lung tissue of Nod2-deficient mice (∼35%) than in that of wild-type mice (∼4%; Fig. 8d). The enhanced inflammation in the respiratory tract of Nod2-deficient mice was also confirmed by higher concentrations of proinflammatory cytokines and chemokines (such as tumor necrosis factor, IL-10 and RANTES) in the BAL fluid of infected Nod2-deficient than in that of wild-type mice (Supplementary Fig. 15b–d). A high RSV load has been associated with enhanced apoptosis of airway epithelial cells and infiltrating neutrophil granulocytes, which contributes to the development of lung lesions and injury37. Indeed, in situ apoptosis analysis of lung sections by TUNEL (terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling) showed enhanced apoptosis in the lungs of RSV-infected Nod2-deficient mice relative to that in the lungs of RSV-infected wild-type mice (Fig. 8e).
Notably, RSV-infected Nod2-deficient mice lost considerably more body weight and had diminished survival relative to their wild-type counterparts (Fig. 8f and Supplementary Fig. 16). We also found less IFN-β production in the BAL fluid of influenza A virus–infected Nod2-deficient mice than in that of influenza A virus–infected wild-type mice (Supplementary Fig. 17). These results demonstrated that Nod2 is a critical component of host antiviral defense mechanisms.
Discussion
In the present study we have identified Nod2 as a viral PRR that can sense viral ssRNA to activate interferon production and antiviral defense. Like RLH receptors, Nod2 associated with MAVS to activate IRF3 and promote interferon production. The importance of Nod2 in host defense was evident from the ability of both cells of the immune response (for example, macrophages) and cells not of the immune response (for example, epithelial cells and MEFs) to use Nod2 for interferon production. The in vivo importance of Nod2 in antiviral responses was evident from the enhanced RSV-induced pathogenesis in infected Nod2-deficient mice.
Other PRRs, including RIG-I, may also be involved in activating an antiviral response against paramyxoviruses like RSV20. For some time, the in vivo relevance of RIG-I in antiviral function was not documented because of the embryonic death of most RIG-I-deficient mice38. However, one strain of RIG-I-deficient mice generated by crossing RIG-I-heterozygous mice with ICR outbred mice, followed by intercrossing of the resultant RIG-I-heterozygous mice38, survives to adulthood. These RIG-I-deficient mice show impaired interferon production and enhanced susceptibility to two positive-sense ssRNA viruses: encephalomyocarditis virus and Japanese encephalitis virus38. However, no studies have been done to demonstrate the importance of RIG-I in activating the antiviral host defense apparatus against negative-sense ssRNA viruses (such as paramyxoviruses).
Uninfected mice have low Nod2 expression39, and we have shown that its expression increased after viral infection. Our observation is similar to those of published studies demonstrating that most PRRs (such as RIG-I) have low expression but their expression is higher after pathogen invasion40,41. Similarly, we found that viruses mediated the induction of Nod2 in various cells. Although in our studies we noted induction of Nod2 in virus-infected MEFs, one study has reported that MDP is unable to induce Nod2 expression in wild-type MEFs; this observation may have been due to a defect in MDP transport to the cytoplasm42. Several other studies have shown that Nod2 expression is stimulated by various bacteria and bacterial components40,41. Similarly, expression of RIG-I (ref. 43) and Mda-5 (ref. 38) in uninfected, unstimulated wild-type MEFs is negligible, but treatment of cells with PAMPs results in upregulation of RIG-I expression43. This mechanism of restricting expression of PRRs in unstimulated cells may be critical for preventing uncontrolled inflammation.
Although Nod2 can be activated by MDP7,8,9,10, so far no studies have demonstrated direct binding of MDP to Nod2; it is known only that lack of Nod2 expression results in loss of MDP responsiveness. Thus, MDP could directly interact with Nod2 or associate with a protein or proteins that form a complex with Nod2. In that context, the interaction of viral ssRNA with Nod2 demonstrated here could also be mediated indirectly via 'bridging' proteins. Interaction of the ssRNA genomes of viruses with RIG-I has also been noted before44,45. In addition to Nod2, cryopyrin (Nalp3), another NLR protein, activates the inflammasome and leads to IL-1 production after stimulation with bacterial and viral (influenza A) RNA46,47,48. Although NLRs such as cryopyrin46,47,48 and NLRX1 (ref. 11) serve an important function in innate immunity by activating the inflammasome and inhibiting interferon production, respectively, no studies have determined whether other NLRs, such as Nod2, can directly contribute to antiviral responses by inducing interferon production.
Published studies have shown that transfection of Nod2 alone (in the absence of any stimulant) into wild-type MEFs results in substantial NF-κB activation49. However, we found that overexpression of Nod2 in 293 cells did not induce marked activation of IRF3 in the absence of external stimuli (such as RSV or synthetic or viral ssRNA). Thus, it seems that Nod2-mediated activation of IRF3 is stimulus dependent, whereas RICK activation is stimulus independent. In summary, our findings have demonstrated that in addition to RIG-I, Mda5 and TLRs, Nod2 can also function as a viral PRR and participate in inducing antiviral signaling. Distinct temporal activation of various PRRs may be needed to generate optimal antiviral responses, and various viruses may trigger the induction of different classes of PRRs.
Methods
Viruses and cell culture.
RSV (A2 strain) and VSV were propagated in HeLa and BHK cells, respectively13,18. Influenza A virus (A/PR/8/34 (H1N1)) was grown in the allantoic cavities of 10-day-old embryonated eggs. All viruses were purified by centrifugation (twice) on discontinuous sucrose gradients. A549 and 293 cells were maintained in DMEM supplemented with 10% (vol/vol) FBS, penicillin, streptomycin and glutamine. Primary NHBE cells (from Lonza) were maintained in bronchial epithelial growth medium according to the supplier's instruction.
Luciferase assay.
First, 293 cells were transfected (Lipofectamine 2000; Invitrogen) with 1 μg of various plasmids (HA-Nod2, HA-Nod1, pcDNA6.1, IRF3-luciferase or IFN-β–luciferase) and 100 ng pRL-null–renilla luciferase. The cells were then infected or treated with RSV, ssRNA or CpG DNA. A549 cells were transfected (Lipofectamine 2000; Invitrogen) with 80 nM Nod2-specific siRNA or control siRNA. At 24 h after siRNA transfection, cells were cotransfected with pRL-null–renilla luciferase (100 ng), IRF3-luciferase (1 μg) or IFN-β–luciferase (1 μg). After 24 h, cells were infected with RSV (multiplicity of infection (MOI), 0.5) or were treated with ssRNA40-LyoVec (1 μg/ml; Invivogen) for various times. Luciferase activity was measured with the Dual-Luciferase Reporter Assay system according to the manufacturer's protocol (Promega). Transfection efficiency was normalized by comparison to expression of renilla luciferase. Luciferase units are presented as relative luciferase activity, which represents the â??fold induction' of luciferase activity after subtraction of the background (cells transfected with pRL-renilla-luciferase plasmid only).
RT-PCR.
Primers for detecting the various genes by RT-PCR are in Supplementary Table 1.
Generation of siRNA.
All siRNA was from Qiagen (sequences, Supplementary Table 1). AllStars Negative Control siRNA (proprietary sequence; 1027281; from Qiagen) was used as a negative control. A549 or 293 cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen) and NHBE cells were transfected with siRNA using PrimeFect Primary Cell siRNA Transfection reagent according to the manufacturer's protocol (Lonza).
Viral infection.
A549 or 293 cells were infected with purified RSV (MOI, 0.5) in serum-free antibiotic free Opti-MEM (Gibco). After adsorption for 1.5 h at 37 °C, cells were washed twice with serum containing DMEM and infection was continued for various times in the presence of serum-containing DMEM. MEFs were infected with purified RSV or influenza A (A/PR/8/34 virus) at an MOI of 1 in serum-free, antibiotic-free Opti-MEM.
Coimmunoprecipitation.
After 293 cells were transfected with various tagged constructs, they were infected with RSV. Cell pellets were lysed (in TBS containing 1% (vol/vol) Triton X-100) and sonicated. All lysates were incubated for 12 h at 4 °C with monoclonal anti-HA agarose beads (HA-7; Sigma-Aldrich). Proteins bound to washed anti-HA–agarose were eluted at a pH of 2.8. Eluted proteins were analyzed by immunoblot with anti-GFP (sc-9996; Santa Cruz) or monoclonal anti-HA (HA-7; Sigma).
Immunofluorescence analysis.
Cells plated on four-well glass chamber slides were transfected with various tagged constructs. Cells were then infected for 4 h or 6 h with RSV (MOI, 1). After infection, cells were fixed with 3.7% (wt/vol) formaldehyde and were made permeable and blocked in permeabilization buffer containing 0.2% (vol/vol) Triton X-100 and 3% (wt/vol) BSA and then were incubated for 1 h at 37 °C with monoclonal anti-HA (Sigma), monoclonal anti-Nod2 (2D9; Cayman Chemical) or anti-MAVS (3993; Cell Signaling Technology). The washed cells were then incubated with secondary antibody (Vector Laboratories). Finally, the washed cells were mounted and then imaged with an LSM510 META laser-scanning confocal microscopy (Zeiss).
Interaction of Nod2 with viral ssRNA.
After 293 cells were transfected with HA-Nod2, they were infected with RSV. Lysates were immunoprecipitated for 4 h at 4 °C with anti-HA–agarose. After beads were washed with TBS, Tri reagent (TRIzol; Sigma-Aldrich) was added for isolation of bound RNA. Primers specific for RSV nucleocapsid protein or GAPDH were used for RT-PCR. For the cell-free interaction assay, lysates of 293 cells expressing HA-Nod2 were incubated with HA–agarose beads. HA-Nod2 bound to the beads was incubated for 45 min at 4 °C with RSV ssRNA or total cellular mRNA (isolated with an RNeasy Mini kit; Qiagen). Beads were washed and RNA isolated from the washed beads was amplified with primers described above.
Virus infection of mice.
Animal studies were approved by the Institutional Animal Care and Use Committee of The University of Texas Health Science Center at San Antonio. Pathogen-free C57BL/6 mice and Nod2-deficient mice (C57BL/6J background) 6–8 weeks old were obtained from The Jackson Laboratory. These Nod2-deficient mice were further back-crossed to the C57BL/6 background for a total of eight generations. Genome-wide single-nucleotide polymorphism analysis of these mice (Harlan Laboratories) showed that wild-type and Nod2-deficient mice are genetically identical, except for the Nod2 deletion (data not shown). Mice were anesthetized with inhaled methoxyfluorane and were inoculated intranasally with RSV (5 × 106 PFU per mouse) in 100 μl low-serum Opti-MEM (Invitrogen). Uninfected control mice were 'sham inoculated' with 100 μl Opti-MEM. For another set of studies, mice were infected intranasally with RSV (5 × 108 PFU per mouse) and the survival of infected mice was monitored for 18 d.
TUNEL and MPO assay.
Formalin-fixed lungs were stained with an in situ TUNEL assay kit Dead End Colorimetric TUNEL system (Promega). Lung neutrophil content was assessed by measurement of MPO activity35,36.
Generation of Nod2 mutants.
Nod2 cDNA was cloned into the pcDNA6-Myc-His vector (Invitrogen) and deletion mutants of Nod2 were constructed by PCR.
Treatment with synthetic and viral ssRNA.
Cells were treated with synthetic ssRNA (1 μg/ml) already conjugated with the transfection reagent (ssRNA40-LyoVec; Invivogen). For isolation of viral ssRNA, purified RSV virion particles were centrifuged for 4 h at 28,000 r.p.m. with an SW32Ti rotor. The ssRNA genome was isolated from the viral pellet with the RNeasy Mini kit. Cells were transfected with viral ssRNA using Lipofectamine 2000 (Invitrogen).
Isolation of MEFs and macrophages.
Alveolar macrophages were collected by centrifugation of BAL fluid at 1,300g for 10 min at 4 °C. After being washed, cell pellets were seeded in 24-well plates. MEFs were prepared as described50. BMMs were obtained from femurs and tibias of wild-type and Nod2-deficient mice and were cultured for 6–8 d.
ELISA.
Human or mouse IFN-β-specific ELISA kits (PBL InterferonSource) were used for ELISA.
Change history
25 June 2010
In the version of this article initially published, some panels in Figure 8e were incorrect. The error has been corrected in the HTML and PDF versions of the article.
References
Kawai, T. & Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137 (2006).
Bose, S. & Banerjee, A.K. Innate immune response against nonsegmented negative strand RNA viruses. J. Interferon Cytokine Res. 23, 401–412 (2003).
Stark, G.R., Kerr, I.M., Williams, B.R.G., Silverman, R.H. & Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 67, 227–264 (1998).
Uematsu, S. & Akira, S. Toll-like receptors and type I interferons. J. Biol. Chem. 282, 15319–15323 (2007).
O'Neill, L.A. How Toll-like receptors signal: what we know and what we don't know. Curr. Opin. Immunol. 18, 3–9 (2006).
Basler, C.F. & Garcia-Sastre, A. Sensing RNA virus infections. Nat. Chem. Biol. 3, 20–21 (2007).
Martinon, F. & Tschopp, J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 26, 447–454 (2005).
Fritz, J.H., Ferrero, R.L., Philpott, D.J. & Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7, 1250–1257 (2006).
Kanneganti, T.D., Lamkanfi, M. & Núñez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Franchi, L., Warner, N., Viani, K. & Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128 (2009).
Moore, C.B. et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451, 573–577 (2008).
Tattoli, I. et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 9, 293–300 (2008).
Kota, S. et al. Role of human β defensin-2 during tumor necrosis factor-α/NF-κB mediated innate anti-viral response against human respiratory syncytial virus. J. Biol. Chem. 283, 22417–22429 (2008).
Voss, E. et al. NOD2/CARD15 mediates induction of the antimicrobial peptide human β-defensin-2. J. Biol. Chem. 281, 2005–2011 (2006).
Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 344, 1917–1928 (2001).
Falsey, A.R., Hennessey, P.A., Formica, M.A., Cox, C. & Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352, 1749–1759 (2005).
Bose, S., Malur, A. & Banerjee, A.K. Polarity of human parainfluenza virus type 3 infection in polarized human lung epithelial A549 cells: Role of microfilament and microtubule. J. Virol. 75, 1984–1989 (2001).
Bose, S., Kar, N., Maitra, R., DiDonato, J.A. & Banerjee, A.K. Temporal activation of NF-κB regulates an interferon-independent innate antiviral response against cytoplasmic RNA viruses. Proc. Natl. Acad. Sci. USA 100, 10890–10895 (2003).
Jamaluddin, M. et al. IFN-β mediates coordinate expression of antigen-processing genes in RSV-infected pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L248–L257 (2001).
Liu, P. et al. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 81, 1401–1411 (2007).
Inohara, N., Ogura, Y. & Nuñez, G. Nods: a family of cytosolic proteins that regulate the host response to pathogens. Curr. Opin. Microbiol. 5, 76–80 (2002).
Franchi, L. et al. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8 (2008).
Wathelet, M.G. et al. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol. Cell 1, 507–518 (1998).
Jafri, H.S. et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J. Infect. Dis. 189, 1856–1865 (2004).
Bolger, G. et al. Primary infection of mice with high titer inoculum respiratory syncytial virus: characterization and response to antiviral therapy. Can. J. Physiol. Pharmacol. 83, 198–213 (2005).
Guerrero-Plata, A. et al. Activity and regulation of α interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 79, 10190–10199 (2005).
Guerrero-Plata, A., Casola, A. & Garofalo, R.P. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J. Virol. 79, 14992–14997 (2005).
Pletneva, L.M., Haller, O., Porter, D.D., Prince, G.A. & Blanco, J.C. Induction of type I interferons and interferon-inducible Mx genes during respiratory syncytial virus infection and reinfection in cotton rats. J. Gen. Virol. 89, 261–270 (2008).
Chávez-Bueno, S. et al. Respiratory syncytial virus-induced acute and chronic airway disease is independent of genetic background: an experimental murine model. Virol. J. 2, 46 (2005).
Castro, S.M. et al. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 174, 1361–1369 (2006).
Hippenstiel, S., Opitz, B., Schmeck, B. & Suttorp, N. Lung epithelium as a sentinel and effector system in pneumonia--molecular mechanisms of pathogen recognition and signal transduction. Respir. Res. 7, 97 (2006).
Yasui, K. et al. Neutrophil-mediated inflammation in respiratory syncytial viral bronchiolitis. Pediatr. Int. 47, 190–195 (2005).
Wang, S.Z. & Forsyth, K.D. The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology 5, 1–10 (2000).
Wang, S.Z. et al. Neutrophils induce damage to respiratory epithelial cells infected with respiratory syncytial virus. Eur. Respir. J. 12, 612–618 (1998).
Bubeck, S.S., Cantwell, A.M. & Dube, P.H. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect. Immun. 75, 697–705 (2007).
Wilmott, R.W., Kitzmiller, J.A., Fiedler, M.A. & Stark, J.M. Generation of a transgenic mouse with lung-specific overexpression of the human interleukin-1 receptor antagonist protein. Am. J. Respir. Cell Mol. Biol. 18, 429–434 (1998).
Welliver, T.P. et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 195, 1126–1136 (2007).
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Opitz, B. et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J. Biol. Chem. 279, 36426–36432 (2004).
Matikainen, S. et al. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J. Virol. 80, 3515–3522 (2006).
Le Goffic, R., Pothlichet, J., Vitour, D., Fujita, T., Meurs, E., Chignard, M. & Si-Tahar, M. Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 178, 3368–3372 (2007).
Abbott, D.W. et al. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol. Cell. Biol. 27, 6012–6025 (2007).
Wang, J. et al. Retinoic acid-inducible gene-I mediates late phase induction of TNF-α by lipopolysaccharide. J. Immunol. 180, 8011–8019 (2008).
Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 (2006).
Kanneganti, T.D. et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006).
Kanneganti, T.D. et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568 (2006).
Ichinohe, T., Lee, H.K., Ogura, Y., Flavell, R. & Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87 (2009).
Kobayashi, K. et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416, 194–199 (2002).
Tominaga, K. et al. MRG15 regulates embryonic development and cell proliferation. Mol. Cell. Biol. 25, 2924–2937 (2005).
Acknowledgements
We thank K. Li (University of Tennessee Health Science Center) for reagents; A. Garcia-Sastre (Mount Sinai School of Medicine) for influenza A virus; Z.J. Chen (University of Texas Southwestern Medical Center) for MAVS-deficient MEFs; the Core Optical Imaging Facility (University of Texas Health Science Center at San Antonio) for confocal images; and K. Moncada Gorena and C. Thomas in the Flow Cytometry Core Facility (supported by the National Institutes of Health (P30 CA54174 to the San Antonio Cancer Institute; P30 AG013319 to the Nathan Shock Center; and P01AG19316)) for flow cytometry. Supported by National Institutes of Health (AI069062 to S.B., CA129246 to S.B. and T32-DE14318 to A.S. and AI067716 to P.H.D.) and the American Lung Association (RG-49629-N to S.B. and AI067716 to P.H.D.).
Author information
Authors and Affiliations
Contributions
A.S. and S.B. designed the experiments and prepared the manuscript; A.S. and T.H.C. did the experiments; R.H. provided technical assistance and did several experiments; Y.X. did the experiments with vaccinia virus; V.F. did the immunofluorescence analysis; K.T. did the studies with mouse embryo fibroblasts; and P.H.D. did the MPO assay.
Corresponding author
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–17 and Supplementary Table 1 (PDF 1608 kb)
Rights and permissions
About this article
Cite this article
Sabbah, A., Chang, T., Harnack, R. et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol 10, 1073–1080 (2009). https://doi.org/10.1038/ni.1782
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.1782
This article is cited by
-
Origins and diversification of animal innate immune responses against viral infections
Nature Ecology & Evolution (2023)
-
Innate immune sensing of pathogens and its post-transcriptional regulations by RNA-binding proteins
Archives of Pharmacal Research (2023)
-
Emerging role of RNA sensors in tumor microenvironment and immunotherapy
Journal of Hematology & Oncology (2022)
-
Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy
Journal of Hematology & Oncology (2022)
-
Thrombosis in COVID-19 infection: Role of platelet activation-mediated immunity
Thrombosis Journal (2021)
