
Abstract
Understanding of the genetic regulation of puberty timing has come largely from studies of rare disorders and population-based studies in women. Here, we report the largest genomic analysis for puberty timing in 55,871 men, based on recalled age at voice breaking. Analysis across all genomic variants reveals strong genetic correlation (0.74, P=2.7 × 10−70) between male and female puberty timing. However, some loci show sex-divergent effects, including directionally opposite effects between sexes at the SIM1/MCHR2 locus (Pheterogeneity=1.6 × 10−12). We find five novel loci for puberty timing (P<5 × 10−8), in addition to nine signals in men that were previously reported in women. Newly implicated genes include two retinoic acid-related receptors, RORB and RXRA, and two genes reportedly disrupted in rare disorders of puberty, LEPR and KAL1. Finally, we identify genetic correlations that indicate shared aetiologies in both sexes between puberty timing and body mass index, fasting insulin levels, lipid levels, type 2 diabetes and cardiovascular disease.
Similar content being viewed by others

Introduction
Voice breaking describes the drop in resonant frequency due to elongation of the larynx in response to androgen exposure1. It is a distinct developmental milestone that occurs during late puberty in males (typically between Tanner stages 3 to 4), and age at voice breaking therefore represents a non-invasive marker for the study of puberty timing in men2.
Age at menarche, the onset of the first menstrual bleed, is a similar marker of pubertal timing in females and has been more widely studied3. Age at menarche in women has been associated with a wide range of disease risks, and previous genome-wide association studies (GWASs) have reported over 100 common loci and five low-frequency coding variants, implicating several previously unsuspected mechanisms involved in the regulation of puberty timing, including post-transcriptional microRNA repression, gamma-aminobutyric acid-B (GABA-B) receptor signalling and nuclear hormone signalling4,5,6,7. Those menarche loci were reportedly enriched for variants also associated with puberty timing in boys, but those analyses were limited by the small number of boys with assessment of physical characteristics of puberty5,6.
Consequently, our understanding of the regulation of puberty timing in males is derived in large part from studies of rare disorders of puberty. To date, ∼20 genes have been implicated in abnormally delayed or absent puberty, including normosmic or anosmic hypogonadotrophic hypogonadism (Kallmann syndrome), while only three genes have been implicated in precocious puberty8,9. Here, we report the first large-scale GWAS of puberty timing in males, based on recalled age at voice breaking in men in the 23andMe study10. By combination with data on recalled age at menarche in women, we show that the genetic architecture of puberty timing has a substantial shared component between males and females, which also overlaps the genetic basis of several health-related traits and diseases.
Results
Genome-wide association signals for age at voice breaking
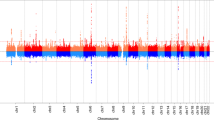
We identified 11 independent genome-wide significant (P<5 × 10−8) signals for age at voice breaking in men located at nine genomic loci (Table 1). Of these, nine signals (mapping to seven loci: in/near LIN28B, MKL2, BSX, TMEM38B, NR4A2, IGSF1 and ALMS1) are correlated (r2>0.05) with reported loci for age at menarche in women5,7. The two novel signals for puberty timing are located in/near LEPR and KAL1, both of which are disrupted in rare disorders of puberty8. The strongest common signal (minor allele frequency (MAF)>5%) for voice breaking was at the LIN28B locus (rs9391253, P=8 × 10−24) where three independent signals were identified (Table 1), which is consistent with the allelic heterogeneity at this locus reported for age at menarche5. All signals are common variants, except for a rare (MAF=1%) intergenic variant near ALMS1, which is associated with 0.32 year per allele later age at voice breaking, and is highly correlated with a rare non-synonymous variant in ALMS1 (rs45501594, T3542S, r2=0.83) that is reportedly associated with age at menarche in women7.
Genetic overlap between puberty timing in men and women
To estimate the shared genetic aetiology between timing of puberty in men and women, we used LD Score Regression11 to calculate the genome-wide genetic correlation (rg) between age at voice breaking in men and age at menarche in women. The observed strong positive correlation (rg=0.74, P=2.7 × 10−70) indicates that many variants have similar influences on puberty timing in males and females. This sex concordance is illustrated by comparison of the effect sizes of all genome-wide significant loci for age at menarche/voice breaking (Fig. 1). Only six of the 123 reported age at menarche loci show significant sex-discordant effects on puberty timing (Pheterogeneity<4.0 × 10−4); these were rs889122-OLFM2, rs466639-RXRG, rs2688325-CSMD1, rs1254337-SIX6, rs6555855-SLIT3 and rs9321659-SIM1/MCHR2. Notably, at the SIM1/MCHR2 locus the reported menarche-age raising allele is associated with younger age at voice breaking in men (P=7.9 × 10−5, Pheterogeneity=1.6 × 10−12).

Dots indicate newly identified voice breaking or previously reported age at menarche variants. NS, not significant.
Confirmation of novel puberty timing signals
Given the strong overall genetic correlation between puberty timing loci in men and women, and the paucity of male puberty timing data available in similarly sized studies, we sought confirmation of novel puberty timing loci identified in men using GWAS data on age at menarche in women (from a combination of publicly available HapMap2-imputed data from 182,416 women in the ReproGen consortium5 and 1,000-Genomes-imputed data from 76,831 additional women in the 23andMe study10,12).
Both of the novel genome-wide significant signals for age at voice breaking in men (in/near LEPR and KAL1) show directionally concordant associations with age at menarche in women (Table 1, LEPR P=1.85 × 10−5; KAL1 2.4 × 10−4). However, the LEPR signal (rs140410685 a 3 bp indel) shows a threefold larger effect on puberty timing in men than in women (Pheterogeneity=3.7 × 10−5).
We next attempted to confirm sub-genome-wide significant signals for age at voice breaking (5 × 10−8<P<1 × 10−6). Of the five signals at this threshold, three were strongly correlated with reported signals for age at menarche5, showing directionally concordant associations with puberty timing in both sexes (rs10980922-ZNF483, rs2282752-WDR6 and rs6681737-NR5A2). The other two signals (rs9350100-RNF144B/ID4 and rs6560352-RORB) showed directionally concordant associations with age at menarche (P<0.01), but with significant heterogeneity between sexes (Table 1). Given this heterogeneity we meta-analysed the two estimates in a random-effects model. Variants at the novel RNF144B-ID4 locus reached genome-wide significance for puberty timing in men and women combined. Although rs6560352 did not reach significance in this meta-analysis (P=2.1 × 10−6), an uncorrelated variant (rs4237264, r2∼0) at this RORB locus was previously reported as a possible signal for age at menarche (P=9 × 10−6)5. In a combined meta-analysis, we robustly confirmed rs4237264 as a novel signal for puberty timing in men and women (P=2 × 10−11).
RORB encodes retinoic acid receptor (RAR)-related orphan receptor beta; notably RORA and RXRG (encoding retinoid X receptor gamma) were previously implicated in age at menarche5. We therefore tested single nucleotide polymorphisms (SNPs) within 500 kb of the remaining six RAR, RAR-related and retinoid X receptor (RXR) encoding genes for associations with puberty timing in our pooled sample of men and women, identifying one additional novel signal ∼350 kb downstream of RXRA (rs416390, P=2 × 10−8) and a further suggestive signal ∼330 kb upstream of RXRB (rs241438, P=5 × 10−6). In aggregate, SNPs in or near a reported list of nuclear hormone receptor genes that contain these RAR-related and RXR genes are significantly enriched for associations with age at voice breaking in men (P=7 × 10−3), as reported for age at menarche in women5.
Genetic correlation between puberty timing and other traits
To inform the likely aetiological relevance of puberty timing in men and women to other health-related outcomes, we used LD Score Regression to test their genetic correlations (rg) with 27 other traits or complex diseases. There is no significant heterogeneity observed in genetic correlations between men and women. In men and women combined, significant genetic correlations (conservatively adjusted for multiple testing: P<1.85 × 10−3 (=0.05/27)) are observed between puberty timing and nine other traits or disease outcomes (Table 2). The strongest genetic correlation is with body mass index (BMI; rg=−0.34, P=4.6 × 10−104); further inverse genetic correlations are observed with polycystic ovary syndrome, fasting insulin levels, type 2 diabetes, triglyceride levels, cardiovascular disease and bone mineral density; and positive genetic correlations are observed with high-density lipoprotein cholesterol levels and adult height.
Discussion
We report a large genetic study of puberty timing in males and females. Our findings are the first to quantify the strongly shared genetic basis for puberty timing between sexes, and this is consistent with the largely sex-concordant effects of disruptive mutations in rare disorders of puberty8,9. Accordingly, our findings support the validity of recalled age at voice breaking in men as a marker of puberty timing in epidemiological studies, consistent with the informative prospective assessment of this phenotype2.
Independent signals at seven loci previously identified for age at menarche in women5 (including three independent signals at the LIN28B locus) passed the genome-wide statistical significance threshold for age at voice breaking in men (Table 1). These included the strongest reported common and low-frequency signals for age at menarche at LIN28B and ALMS1, respectively, and other signals with relatively large effects. This observation is consistent with the largely shared genetic architecture for puberty timing between sexes.
The overlapping genetic architecture for puberty timing in men and women provided the rationale for a pooled meta-analysis across the sexes. Notably, we identified two novel signals, near RORB and RXRA, which add to the reported signals near to other retinoic acid receptor-related genes, RORA and RXRG5, and a fifth signal (near RXRB) showed sub-genome-wide significant association with puberty timing. The retinoic acid receptor-related and retinoid X receptors function as transcription factors that dimerize and regulate nuclear receptors to influence cell differentiation, development, circadian rhythm and metabolism13. Their receptor partners include the canonical receptors for oestrogen and androgens among other hormones and metabolites14, and their conformational changes may alter receptor sensitivity15. Collectively these findings strengthen the evidence for an aetiological role of retinoic acid and retinoid receptors in the regulation of puberty timing, although their relative importance to male versus female puberty remains to be established. Further studies are also needed to identify functional links between these allelic signals and specific gene and protein functions.
Three additional novel signals reached genome-wide significance for puberty timing, represented by variants in/near RNF144B-ID4, LEPR and KAL1. All three were associated with puberty timing in both men and women, two of which had stronger effects in males (LEPR and RNF144B-ID4). The signal at 6p22.3 resides in a gene desert with the two nearest genes, ID4 and RNF144B, located 760 kb and 607 kb away, respectively. Notably, it also lies 852 kb from KDM1B, a gene in the same family of histone demethlyases highlighted previously for age at menarche (variants in/near KDM3B, KDM4A and KDM4C represented genome-wide significant signals)5. Variants near KAL1 on Xp22.31 had not been highlighted by previous GWAS of female puberty timing due to paucity of X-chromosome data in those studies. rs5978985 is correlated with a reported signal for circulating free testosterone concentrations in males (r2=0.35 with rs5934505)16 and KAL1 encodes the extracellular matrix glycoprotein anosmin-1 implicated in the embryonic migration of gonadotrophin releasing hormone and olfactory neurons. Deleterious mutations in KAL1 cause X-linked Kallmann syndrome, characterized by hypogonadotropic hypogonadism and anosmia8. rs140410685 near LEPR, which encodes the leptin receptor, is uncorrelated with a reported neighbouring signal for age at menarche (r2∼0 with rs10789181)5, and shows no reported association with adult BMI in reported GWAS meta-analyses (rs2186245 r2=1 proxy, PBMI=0.33, N=233,888)17.
In rare disorders of puberty, disruptive mutations usually have similar effects on puberty timing in both sexes8,9. Notable exceptions are rare mutations in the genes that encode the pituitary hormones, follicle-stimulating hormone and luteinizing hormone, which disrupt puberty in females but not in males18. In normal populations, rapid postnatal weight gain predicts earlier puberty timing in both sexes, but the influence of low birth weight is apparent only in females2. We found that only a small minority of signals showed sex-discordant effects, most notably rs9321659 at the SIM1-MCHR2 locus. SIM1 encodes a transcription factor regulator of hypothalamic paraventricular nucleus development and function. Rare deleterious mutations cause hyperphagia and severe early onset obesity affecting both males and females19; however, rs9321659 is reportedly not associated with adult BMI (P=0.56)17. Overall, these data suggest that combining male and female data genome wide is likely to yield novel shared puberty loci, but care should be taken to assess for potential heterogeneity in the effects.
Finally, our observed genetic correlations indicate the relevance of puberty timing to later life health outcomes in both men and women. Consistent with traditional epidemiological evidence20,21,22, earlier puberty timing was genetically related to higher risks of adverse health-related outcomes, including higher BMI, polycystic ovary syndrome, type 2 diabetes, lipid profiles and cardiovascular disease; it was favourable only for bone mineral density. LD score regression is a powerful tool to identify potential causal relationships between traits; however, limitations include the inability to establish causal directions and to adjust for potential-mediating factors. The relationship between puberty timing and obesity risk is complex, with plausible bi-directional mechanisms. Previous studies have reported that genome-wide significant signals for higher BMI, both in combination and individually, are associated with earlier puberty timing in females5,23,24. Conversely, in the opposite direction, earlier puberty timing associated with the LIN28B rs314276 C-allele leads to faster adolescent weight gain and higher post-pubertal BMI, without effects on pre-pubertal BMI25. Further studies are required to explore whether health outcomes related to earlier puberty timing are fully mediated by higher BMI.
In summary, this large-scale assessment of the genetic architecture of puberty timing in males quantifies the extent of shared aetiology between sexes, extends the evidence implicating retinoic acid-related receptors in the regulation of puberty timing, and supports the relevance of puberty timing in both sexes to the aetiologies of various health-related outcomes.
Methods
Genome-wide association study for age at voice breaking
Genome-wide SNP data were generated from one or more of three genotyping arrays in up to 55,871 men aged 18 or older of European ancestry from the 23andMe study10,12, who reported their recalled age at voice breaking, by online questionnaire in response to the question ‘How old were you when your voice began to crack/deepen?’ Participants answered into one of the predefined age bins (under 9, 9–10 years old, 11–12 years old, 13–14 years old, 15–16 years old, 17–18 years old, 19 years old or older), scored from 0 to 6. Genetic effect estimates from these 2-year bins were re-scaled to 1-year effect estimates post analysis. We previously validated the accuracy of this approach by comparing re-scaled 2-year estimates for age at menarche (recorded in the same way) to those obtained from studies recording age at menarche by year. No significant heterogeneity was detected across these two approaches for known menarche loci7. 23andMe participants provided informed consent to take part in this research under a protocol approved by Ethical and Independent Review Services, an institutional review board accredited by the Association for the Accreditation of Human Research Protection Programs. Before imputation, we excluded SNPs with Hardy–Weinberg equilibrium P<10−20, call rate <95%, or with large allele frequency discrepancies compared with European 1,000 Genomes reference data. Frequency discrepancies were identified by computing a 2 × 2 table of allele counts for European 1,000 Genomes samples and 2,000 randomly sampled 23andMe participants with European ancestry, and identifying SNPs with a χ2 P<10−15. Genotype data were imputed against the March 2012 ‘v3’ release of 1,000 Genomes reference haplotype panel. Genetic association results were obtained from linear regression models assuming additive allelic effects. These models included as covariates—age and the top five genetically determined principal components to account for population structure. The reported SNP association test P values were computed from likelihood ratio tests. Results were further adjusted for a lambda GC value of 1.069 to correct for any residual test statistic inflation due to population stratification. LD score regression analysis26 also confirmed that principal component correction had appropriately controlled for inflation due to population stratification (pre-GC calculated intercept∼1) before the more conservative GC correction.
Independent signals were identified using a combination of distance-based clumping and approximate conditional analysis. Firstly, regions were defined on the basis of physical proximity, with the most strongly associated SNP representing the association signal for that region. We then tested for the presence of multiple statistically independent signals in each region using approximate conditional analysis implemented in GCTA27. Independent signals indicated by SNP P values <1 × 10−6 were considered in follow-up analyses. A signal was considered to be the same as a previously reported menarche locus if it had a pairwise r2>0.05.
Combined analyses with other puberty data sets
We followed up these selected SNPs in two additional sources of data: reported publicly available HapMap2 reference panel-imputed GWAS results for age at menarche from 182,416 women in the ReproGen consortium5; and independent 1,000 genomes reference panel-imputed GWAS data for age at menarche in 76,831 women in the 23andMe study7,10.
These additional samples were considered in three analytical designs. Firstly, all data in women (n=259,247) were combined to estimate effects of the individual selected SNPs on age at menarche, using fixed-effects inverse variance-weighted meta-analysis with all effect estimates reported on a per year scale. Secondly, for genetic correlation analyses with age at menarche, women in ReproGen consortium cohorts genotyped by the custom ‘iCOGs’ array were excluded to obtain a consistent sample size across GWAS SNPs (leaving data for analysis on 209,820 women). Thirdly, all GWAS results for puberty timing in men and women (n=315,118) were combined using inverse variance fixed-effects meta-analysis. LD Score Regression11,26 showed that combining GWAS data from men and women did not introduce substantial test statistic inflation due to possible relatedness between strata (cross-trait intercept 0.016, s.e. 0.005). Heterogeneity between men and women for individual SNP associations was quantified by the I2 statistic generated by METAL software. SNPs that demonstrated heterogeneity were analysed in a random-effects model implemented by Han and Eskin28.
Genetic correlations
Genetic correlations (rg) were calculated between age at voice breaking in men, age at menarche in women, and 27 other complex traits/diseases in publicly available data sets using LD Score Regression11,26. Data sets used can be downloaded from http://www.med.unc.edu/pgc/downloads. Sample numbers in the studies of each trait are shown in Table 2. The only non-publicly available data set used was a polycystic ovary syndrome GWAS performed on 5,184 self-reported cases and 82,759 controls from the 23andMe study29. A conservative Bonferroni corrected P value threshold of 0<1.85 × 10−3 (=0.05/27) was used to define significant associations.
Additional information
How to cite this article: Day, F. R. et al. Shared genetic aetiology of puberty timing between sexes and with health-related outcomes. Nat. Commun. 6:8842 doi: 10.1038/ncomms9842 (2015).
References
Harries, M. L., Walker, J. M., Williams, D. M., Hawkins, S. & Hughes, I. A. Changes in the male voice at puberty. Arch. Dis. Child. 77, 445–447 (1997).
Ong, K. K. et al. Timing of voice breaking in males associated with growth and weight gain across the life course. J. Clin. Endocrinol. Metab. 97, 2844–2852 (2012).
Parent, A. S. et al. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr. Rev. 24, 668–693 (2003).
Elks, C. E. et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat. Genet. 42, 1077–1085 (2010).
Perry, J. R. et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 514, 92–97 (2014).
Ong, K. K. et al. Genetic variation in LIN28B is associated with the timing of puberty. Nat. Genet. 41, 729–733 (2009).
Lunetta, K. L. et al. Rare coding variants and X-linked loci associated with age at menarche. Nat. Commun. 6, 7756 (2015).
Silveira, L. F. & Latronico, A. C. Approach to the patient with hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 98, 1781–1788 (2013).
Abreu, A. P. et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 368, 2467–2475 (2013).
Hinds, D. A. et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat. Genet. 45, 907–911 (2013).
Bulik-Sullivan, B. K. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. (in the press) doi:10.1038/ng.3406 (2015).
Tung, J. Y. et al. Efficient replication of over 180 genetic associations with self-reported medical data. PLoS ONE 6, e23473 (2011).
Dawson, M. I. & Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 1821, 21–56 (2012).
Chuang, K. H. et al. 9-cis-retinoic acid inhibits androgen receptor activity through activation of retinoid X receptor. Mol. Endocrinol. 19, 1200–1212 (2005).
Macoritto, M. et al. Phosphorylation of the human retinoid X receptor alpha at serine 260 impairs coactivator(s) recruitment and induces hormone resistance to multiple ligands. J. Biol. Chem. 283, 4943–4956 (2008).
Ohlsson, C. et al. Genetic determinants of serum testosterone concentrations in men. PLoS Genet. 7, e1002313 (2011).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Day, F. R., Perry, J. R. & Ong, K. K. Genetic regulation of puberty timing in humans. Neuroendocrinology (in the press) doi: 10.1159/000431023 (2015).
Ramachandrappa, S. et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J. Clin. Invest. 123, 3042–3050 (2013).
Day, F., Elks, C. E., Murray, A. M., Ong, K. K. & Perry, J. R. Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the UK Biobank study. Sci. Rep. 5, 11208 (2015).
Prentice, P. & Viner, R. M. Pubertal timing and adult obesity and cardiometabolic risk in women and men: a systematic review and meta-analysis. Int. J. Obes. (Lond) 37, 1036–1043 (2013).
Elks, C. E. et al. Age at menarche and type 2 diabetes risk: the EPIC-InterAct study. Diabetes Care 36, 3526–3534 (2013).
Mumby, H. S. et al. Mendelian randomisation study of childhood BMI and early menarche. J. Obes. 2011, 180729 (2011).
Cousminer, D. L. et al. Genome-wide association study of sexual maturation in males and females highlights a role for body mass and menarche loci in male puberty. Hum. Mol. Genet. 23, 4452–4464 (2014).
Ong, K. K. et al. Associations between the pubertal timing-related variant in LIN28B and BMI vary across the life-course. J. Clin. Endocrinol. Metab. 96, E125–E129 (2011).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Day, F. R. et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat. Commun 6, 8464 (2015).
Acknowledgements
This work was supported by the Medical Research Council [U106179472; MC_U106179472; U106179471; MC_U106179471] and the National Human Genome Research Institute of the National Institutes of Health (grant number R44HG006981 to 23andMe). We thank the research participants and employees of 23andMe for making this work possible. HKF is supported by the Fannie and John Hertz Foundation.
Author information
Authors and Affiliations
Contributions
All authors read and contributed to each version of the manuscript. F.R.D., B.B.-S., H.K.F. and J.R.B.P. carried out analysis. D.A.H., J.M.M. and J.Y.T. performed genotyping and phenotyping. D.A.H., J.Y.T., K.K.O. and J.R.B.P. contributed to study design and project management.
Corresponding author
Ethics declarations
Competing interests
D.A.H. and J.Y.T. are employees of and own stock or stock options in 23andMe, Inc. The remaining authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Day, F., Bulik-Sullivan, B., Hinds, D. et al. Shared genetic aetiology of puberty timing between sexes and with health-related outcomes. Nat Commun 6, 8842 (2015). https://doi.org/10.1038/ncomms9842
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms9842
This article is cited by
-
DNA methylation sites in early adulthood characterised by pubertal timing and development: a twin study
Clinical Epigenetics (2023)
-
Analysis of transcriptional changes in the immune system associated with pubertal development in a longitudinal cohort of children with asthma
Nature Communications (2023)
-
COVID-19 lockdown and the rate of central precocious puberty
Journal of Endocrinological Investigation (2023)
-
Prenatal Exposure to Parental Lifestyle Factors, Diseases, and Use of Medications and Male Pubertal Development: a Review of Epidemiological Studies Published 2017–2022
Current Epidemiology Reports (2023)
-
The Association Between Puberty Timing and Body Mass Index in a Longitudinal Setting: The Contribution of Genetic Factors
Behavior Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
