
Abstract
More than 100 loci have been identified for age at menarche by genome-wide association studies; however, collectively these explain only ∼3% of the trait variance. Here we test two overlooked sources of variation in 192,974 European ancestry women: low-frequency protein-coding variants and X-chromosome variants. Five missense/nonsense variants (in ALMS1/LAMB2/TNRC6A/TACR3/PRKAG1) are associated with age at menarche (minor allele frequencies 0.08–4.6%; effect sizes 0.08–1.25 years per allele; P<5 × 10−8). In addition, we identify common X-chromosome loci at IGSF1 (rs762080, P=9.4 × 10−13) and FAAH2 (rs5914101, P=4.9 × 10−10). Highlighted genes implicate cellular energy homeostasis, post-transcriptional gene silencing and fatty-acid amide signalling. A frequently reported mutation in TACR3 for idiopathic hypogonatrophic hypogonadism (p.W275X) is associated with 1.25-year-later menarche (P=2.8 × 10−11), illustrating the utility of population studies to estimate the penetrance of reportedly pathogenic mutations. Collectively, these novel variants explain ∼0.5% variance, indicating that these overlooked sources of variation do not substantially explain the ‘missing heritability’ of this complex trait.
Similar content being viewed by others

Introduction
Age at menarche, the onset of first menstruation in females, indicates the start of reproductive maturity and is a commonly reported marker of pubertal timing. One hundred and six genomic loci for this highly heritable trait have been mapped by genome-wide association studies (GWAS), implicating many previously unsuspected mechanisms1. However, to date that approach has been limited to consideration of only those genetic variants captured by autosomal HapMap2 reference panels. In particular, like most GWAS for other complex traits, previous GWAS for age at menarche provided poor coverage for low-frequency variants and omitted sex chromosome data.
Here we report a dual strategy for assessing genetic variation overlooked by those prior efforts: low-frequency protein-coding variants genotyped by large-scale exome-focussed arrays and high-density X-chromosome single-nucleotide polymorphism (SNP) genotyping and imputation. We identify several new associations between rare protein-coding and X-linked variants with age at menarche in women of European ancestry. The findings implicate new mechanisms that regulate puberty timing, but collectively these novel variants explained only ∼0.5% of the variance, indicating that these often overlooked sources of variation that do not substantially explain the ‘missing heritability’ of this complex trait.
Results
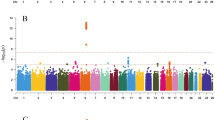
From the exome array studies, 61,734 low-frequency (minor allele frequency (MAF) <5%) variants passed quality-control (QC) criteria in a combined sample of up to 76,657 women of European ancestry from 19 studies (Supplementary Table 1). Gene-based burden and SKAT tests that aggregate the effects of variants with MAF<1% yielded no significant associations with age at menarche. A linear regression test was used to derive all P values obtained in this study. Meta-analysis of individual variant associations with questionnaire-reported variation in age at menarche (restricted to the ages of 9–17 years) in this discovery phase identified one signal at genome-wide statistical significance (P<5 × 10−8); this was a rare missense variant in the Alström’s syndrome gene (ALMS1, rs45501594, p.T3544S, MAF 1%; P=4.6 × 10−10). For follow-up testing in up to 116,317 independent women of European ancestry from the deCODE (Diabetes Epidemiology: Collaborative analysis of Diagnostic criteria in Europe) and 23andMe studies, we selected rs45501594 and 23 other variants that met the following criteria: protein coding, present in over half of the exome array studies, and with association P<5 × 10−4. In the follow-up samples, 7 of the 20 variants that passed QC showed directionally concordant confirmatory associations with P<0.05, of which five reached genome-wide significance in a combined meta-analysis of discovery phase and follow-up data (Table 1, Fig. 1, Supplementary Fig. 1). No significant heterogeneity between studies was observed at any of these loci (Supplementary Fig. 2).

Test statistics are shown from the exome-chip discovery-phase samples, with the exception of the five labelled loci that indicate results from the combined discovery and replication set.
The rare missense variant in ALMS1 (rs45501594, Supplementary Fig. 3) remained the strongest signal identified using exome array studies (combined: P=6.8 × 10−20). In the follow-up samples, each rare allele was associated with 0.23-year-later age at menarche, an effect size more than double that of any genetic variant previously reported for puberty timing in the general population. This strong signal was not detected by the previous HapMap2-based GWAS as it is poorly tagged by common SNPs in that reference panel (maximum proxy SNP, r2=0.24). Deleterious mutations in this gene cause Alström’s syndrome (OMIM no. 203800), a rare, autosomal-recessive disorder characterized by cone–rod dystrophy, sensorineural hearing loss, dilated cardiomyopathy, childhood obesity, insulin resistance, diabetes mellitus, hypogonadotropic hypogonadism in males, menstrual irregularities and early puberty in females, and short stature in adulthood2. Hypogonadism was also invariably observed in an ALMS1 gene-trapped mouse model3.
The variant with largest effect was a rare stop-gain mutation in the tachykinin receptor 3 gene (TACR3; rs144292455, MAF=0.08%, combined P=2.8 × 10−11, Supplementary Fig. 3); in follow-up samples each rare allele was associated with 1.25-year-later age at menarche. Common HapMap2 SNPs at the TACR3 locus were previously associated with age at menarche1; however, the rare variant rs144292455 is not tagged by the HapMap2 or conventional 1000G imputation (it was directly genotyped in 23andMe and was imputed in deCODE). Statistical independence was confirmed by observing significant association with the common TACR3 SNP in a sensitivity analysis within a participating study (Women's Genome Health Study (WGHS), Supplementary Table 1) that excluded rare allele carriers. The rare allele causes a premature stop codon (p.W275X) in the fifth transmembrane segment of the 465 amino-acid receptor for the neuropeptide neurokinin B, and is the most frequently reported TACR3 mutation in the rare reproductive disorder idiopathic hypogonadotropic hypogonadism (idiopathic hypogonadotropic hypogonadism (IHH), OMIM no. 614840)4. Both homozygous and heterozygous p.W275X variants have been reported in male IHH cases with features of ‘early androgen deficiency’; however, notably the heterozygous cases showed evidence of spontaneous neuroendocrine recovery. Our findings suggest that heterozygous p.W275X variants contribute to the normal variation in puberty timing, whereas homozygous inheritance or possibly compound heterozygosity is required for IHH.
A low-frequency missense variant in the LAMB2 gene was associated with 0.08-year-later age at menarche (rs35713889, p.G914R, MAF 4%; P=1.1 × 10−11; Supplementary Fig. 3). In the same region (3p21.31) we previously reported a HapMap2 GWAS locus for age at menarche (locus 19a and 19b in ref. 1); however, the low-frequency variant rs35713889 is poorly tagged by common HapMap2 SNPs (the best proxy rs1134043, r2=0.24, was reportedly not associated with age at menarche: P=0.35 (ref. 1)). The strongest reported1 HapMap2 signal at this locus is only weakly correlated with rs35713889 (rs3870341, MAF=26%; r2=0.07, distance 422 kb), and both signals remained significant when jointly tested in a follow-up sample of 76,831 women from the 23andMe study (in separate models: rs35713889: β=0.08 years per allele, P=0.0001 and rs3870341: β=0.04, P=4.5 × 10−5; in the joint model: rs35713889: β=0.06, P=0.004 and rs3870341: β=0.03, P=0.001). LAMB2 encodes one of 15 subunits of Laminin, an extracellular matrix glycoprotein with a key role in the attachment, migration and organization of cells into tissues during embryonic development. Rare recessive mutations in LAMB2 cause Pierson’s syndrome (OMIM no. 609049), a disorder characterized by congenital nephrotic syndrome and ocular anomalies, typically with microcoria5; neurological abnormalities are also described likely because of cortical laminar disorganization6. Common variants in/near other Laminin genes have been reported for a broad range of complex traits, including type 2 diabetes7, refractive error8, colorectal cancer9, IgG glycosylation10, ulcerative colitis11 and coffee consumption12.
A low-frequency missense variant in the TNRC6A gene was associated with later age at menarche (rs113388806; p.Q1112H; MAF 4.7%; β=0.08 years per allele; P=1.1 × 10−11; Supplementary Fig. 3). This signal was only moderately well tagged by common HapMap2 SNPs (best proxy: rs12447003, r2=0.36, reported association with age at menarche: P=0.0005 (ref. 1)). TNRC6A encodes an Argonaute-navigator protein, responsible for post-transcriptional gene silencing through RNA interference and microRNA pathways13. This finding further extends the range of epigenetic mechanisms implicated in the regulation of puberty14.
A low-frequency missense variant in PRKAG1 was associated with earlier age at menarche (rs1126930; p.T98S; MAF 3.4%; β=−0.09 years per allele, P=9.6 × 10−11; Supplementary Fig. 3). This low-frequency variant is only moderately well tagged by common HapMap2 SNPs (max r2=0.36), which reportedly showed subgenome-wide significant association with age at menarche (rs11837234, P=3.1 × 10−6)1. This PRKAG1 missense variant is in the same locus as, but not correlated to, a reported1 common signal for age at menarche (rs7138803, 848 kb apart, r2=0.02). PRKAG1 encodes the gamma-1 regulatory subunit of AMP-activated protein kinase, which senses and maintains cellular energy homeostasis by promoting fatty-acid oxidation and inhibiting fatty-acid synthesis; PRKAG1 is overexpressed in ovarian carcinomas15 and is somatically mutated in colorectal cancers16.
Our second genotyping approach considered X-chromosome GWAS SNPs in up to 76,831 women of European ancestry from the 23andMe study17. Imputation was performed against the 1000 Genomes reference, yielding genotype data for ∼266,000 X-chromosome variants (MAF>1%). Two signals, in/near IGSF1 and FAAH2, reached genome-wide significance for association with age at menarche and both associations were confirmed in 39,486 independent women of European ancestry from the deCODE study.
Common variants in and near IGSF1 were robustly associated with age at menarche (lead SNP: rs762080, MAF=24%; β=0.06 years per allele, P=9.4 × 10−13; Supplementary Fig. 4). IGSF1 encodes the immunoglobulin superfamily member 1, which is a plasma membrane glycoprotein highly expressed in the pituitary gland and testis. Rare X-linked mutations in IGSF1 were recently described to cause central hypothyroidism, hypoprolactinemia, delayed puberty and macro-orchidism in males (OMIM no. 300888)18,19. Heterozygous female carriers reportedly had normal age at menarche; however, 6/18 had central hypothyroidism and 4/18 underwent oophorectomy for ovarian cysts19.
The second X-chromosome locus, in Xp11.21 (lead SNP rs5914101 is intronic in FAAH2, MAF 24%, β=0.05 years per allele, P=1.9 × 10−10; Supplementary Fig. 4), lies within the critical region for Turner’s syndrome, which is the most common cause of primary ovarian insufficiency20. FAAH2 encodes fatty-acid amide hydrolase 2. This enzyme catalyses the hydrolysis and degradation of bioactive fatty-acid amides, a large class of endogenous signalling lipids including the endocannabinoids, which modulate several physiological processes, including feeding, inflammation, pain, sleep and various reproductive processes, including hypothalamic gonadotropin-releasing hormone secretion21,22.
We sought to further functionally characterize the seven genes implicated by these analyses using expression data on 53 tissue types from the Genotype-Tissue Expression consortium23. All seven genes showed high relative tissue expression in the ovary and/or brain (specifically the hypothalamus; Supplementary Fig. 5); however, none of the lead SNPs showed a significant association with mRNA transcript abundance. None of the identified variants were associated with body mass index in 74,071 adults from the deCODE study (all P>0.05), indicating that their effects on puberty timing are unlikely to be mediated by body mass index.
Discussion
In summary, by large-scale analysis of genetic variation not captured by previous GWAS for age at menarche, we identified several low-frequency exonic variants of relatively large effect and two common X-chromosome signals. The implicated genes provide new insights into the mechanisms that link energy homeostasis to puberty timing, indicate possible roles of RNA-mediated gene silencing and fatty-acid amide signalling, and link genes behind rare autosomal, X-linked and syndromic disorders of puberty to normal variation in reproductive timing. Our findings using dense exome arrays in large unselected populations are informative for the clinical interpretation of heterozygous TACR3 variants in patients with rare disorders. In the deCODE study these novel variants collectively explained only 0.5% of the variance in age at menarche, suggesting that these often overlooked sources of genetic variation do not contribute disproportionately to the missing heritability of this complex trait. While variants with MAF below 1% are likely not well represented here, our findings indicate that, similar to other complex traits24, the genetic architecture of puberty timing is likely dominated by the additive effects of hundreds or even thousands of variants, each with relatively small effect.
Methods
Exome array discovery analysis
Exome array genotype data were generated across 19 studies in up to 76,657 women of genetically determined European ancestry with questionnaire-reported age at menarche between ages 9 and 17 years (Supplementary Table 1). Exome array genotype calling for three studies (Framingham Heart Study (FHS), the Atherosclerosis Risk in Communities (ARIC) and Rotterdam Study (RS); totalling ∼9,000 women) was performed jointly as part of the CHARGE joint calling protocol25, which included over 62,000 individuals. Four additional studies (Cambridge Cancer, KORA, Korcula, Generation Scotland, total N∼9,700) used the cluster file made available by CHARGE to call genotypes. Other studies followed standard calling and QC protocols for the Exome array (Supplementary Table 2). Each contributing study ran a linear regression model on age at menarche, adjusted for birth year and principal components derived from genotypes, using the skatMeta/seqMeta package in R. Studies with family data included a random effect to account for relationships. Alleles were aligned to a common reference file before association testing (SNPInfo_HumanExome-12v1_rev5.tsv.txt available at http://www.chargeconsortium.com/main/exomechip/) and variants with MAF>5% in the meta-analysis were excluded. We performed gene-based testing (within seqMeta) for low-frequency variants using fixed effect burden tests, which assume that all rare variants have the same effect direction and size (scaled by a weight determined by allele frequency), and SKAT tests, which assume that rare variant effects are random and can contain a mixture of null, protective and risk rare alleles. These tests were run using three variant filters, all of which included only variants with MAF<1%: (1) all non-synonymous; (2) non-synonymous annotated as ‘damaging’ (conserved and predicted damaging, see http://www.chargeconsortium.com/main/exomechip/); and (3) only loss of function. The multiple testing adjustment included two tests × three filters × number of genes, requiring study-wise significance threshold P<1.14 × 10−6. For individual variants, a fixed-effects inverse variance-weighted meta-analysis was performed across all studies using METAL (http://www.sph.umich.edu/csg/abecasis/Metal/), with associations considered significant at a conservative genome-wide significance threshold of P<5 × 10−8.
Exome array follow-up studies
We performed follow-up testing of selected exome array variants in the 23andMe study (as described below) and also in 39,486 independent women of European ancestry from the deCODE study, Iceland, who had genotypes on over 34 million variants by imputation of whole-genome sequencing-identified SNPs and indels on Illumina SNP chip data (Supplementary Table 1)26. Variants from both studies were required to either pass genotyping QC (23andMe only, described below) or have imputation quality score >0.4. X-chromosome follow-up was performed in the deCODE study alone. All participants in all published studies provided informed consent, and the research protocol of each study was approved by their local research ethics committee1.
1000G X-chromosome discovery meta-analysis
X-chromosome SNP data were generated in up to 76,831 women of European ancestry from the 23andMe study17,27, with questionnaire-reported age at menarche between the ages of 8 and 16 years, and who were genotyped on one or more of three GWAS arrays that also included customized content on human pathogenic variants (Supplementary Table 1)28. 23andMe participants provided informed consent to take part in this research under a protocol approved by the AAHRPP-accredited institutional review board, Ethical and Independent Review Services. Before imputation, we excluded SNPs with Hardy–Weinberg equilibrium P<10−20, call rate <95% or with large allele frequency discrepancies compared with European 1000 Genomes reference data. Frequency discrepancies were identified by computing a 2 × 2 table of allele counts for European 1000 Genomes samples and 2,000 randomly sampled 23andMe customers with European ancestry, and identifying SNPs with a χ2 P<10−15. Genotype data were imputed against the March 2012 ‘v3’ release of 1000 Genomes reference haplotypes. Age at menarche was assessed by questionnaire and recorded in 2-year-age bins, which were rescaled to 1-year effect estimates post analysis. The validity of this approach was confirmed by the lack of significant heterogeneity between rescaled 23andMe menarche estimates for the 123 previously identified signals and their reported effects1. Association results were obtained from linear regression models assuming additive allelic effects. These models included covariates for age and the top five GWAS SNP principal components to account for residual population structure. Results were further adjusted for a lambda genomic control value of 1.152 to correct for any residual test statistic inflation due to population stratification. Linkage disequilibrium score regression analysis (LDSC)29 confirmed that principle component correction appropriately controlled for potential test statistic inflation due to population stratification (pre-genomic control-corrected calculated intercept ∼1). The reported association test P values were computed from likelihood ratio tests.
X-chromosome follow-up
Identified X-chromosome variants were replicated in 39,486 women from the deCODE study, as described above.
Additional information
How to cite this article: Lunetta, K. L. et al. Rare coding variants and X-linked loci associated with age at menarche. Nat. Commun. 6:7756 doi: 10.1038/ncomms8756 (2015).
Change history
17 December 2015
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
Perry, J. R. et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 514, 92–97 (2014).
Ozanturk, A. et al. The phenotypic and molecular genetic spectrum of Alstrom syndrome in 44 Turkish kindreds and a literature review of Alstrom syndrome in Turkey. J. Hum. Genet. 60, 1–9 (2014).
Collin, G. B. et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum. Mol. Genet. 14, 2323–2333 (2005).
Gianetti, E. et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J. Clin. Endocrinol. Metab. 95, 2857–2867 (2010).
Matejas, V. et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 31, 992–1002 (2010).
Radner, S. et al. beta2 and gamma3 laminins are critical cortical basement membrane components: ablation of Lamb2 and Lamc3 genes disrupts cortical lamination and produces dysplasia. Dev. Neurobiol. 73, 209–229 (2013).
Perry, J. R. et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet. 8, e1002741 (2012).
Verhoeven, V. J. et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat. Genet. 45, 314–318 (2013).
Houlston, R. S. et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat. Genet. 42, 973–977 (2010).
Lauc, G. et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 9, e1003225 (2013).
Consortium, U. I. G.. et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 41, 1330–1334 (2009).
Amin, N. et al. Genome-wide association analysis of coffee drinking suggests association with CYP1A1/CYP1A2 and NRCAM. Mol. Psychiatry. 17, 1116–1129 (2012).
Hafner, M. et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141 (2010).
Lomniczi, A., Wright, H. & Ojeda, S. R. Epigenetic regulation of female puberty. Front. Neuroendocrinol. 36, 90–107 (2014).
Li, C., Liu, V. W., Chiu, P. M., Chan, D. W. & Ngan, H. Y. Over-expressions of AMPK subunits in ovarian carcinomas with significant clinical implications. BMC Cancer 12, 357 (2012).
Seshagiri, S. et al. Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664 (2012).
Hinds, D. A. et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat. Genet. 45, 907–911 (2013).
Sun, Y. et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat. Genet. 44, 1375–1381 (2012).
Joustra, S. D. et al. The IGSF1 deficiency syndrome: characteristics of male and female patients. J. Clin. Endocrinol. Metab. 98, 4942–4952 (2013).
Zinn, A. R. et al. Evidence for a Turner syndrome locus or loci at Xp11.2-p22.1. Am. J. Hum. Genet. 63, 1757–1766 (1998).
Gammon, C. M., Freeman, G. M. Jr, Xie, W., Petersen, S. L. & Wetsel, W. C. Regulation of gonadotropin-releasing hormone secretion by cannabinoids. Endocrinology 146, 4491–4499 (2005).
Meccariello, R., Battista, N., Bradshaw, H. B. & Wang, H. Updates in reproduction coming from the endocannabinoid system. Int. J. Endocrinol. 2014, 412354 (2014).
Consortium, G. T.. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Wood, A. R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014).
Grove, M. L. et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS ONE 8, e68095 (2013).
Steinthorsdottir, V. et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 46, 294–298 (2014).
Tung, J. Y. et al. Efficient replication of over 180 genetic associations with self-reported medical data. PLoS ONE 6, e23473 (2011).
Eriksson, N. et al. Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet. 6, e1000993 (2010).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Acknowledgements
Sources of funding for the contributing studies are listed in Supplementary Table 3.
Author information
Authors and Affiliations
Consortia
Contributions
All authors reviewed the original and revised manuscripts. Analysis: K.L.L., F.R.D., P.S., K.S.R., T.E., C.E.E., E.A., C.H., J.E.H., E.M., E.P., A.R., L.M.R., U.M.S., L.S., A.T., D.J.T., M.T., C.A.W., L.M.Y.-A. and J.R.B.P.; sample collection, phenotyping and genotyping: A.C.A., C.B., A.D.C., F.C., E.W.D., A.M.D., I.G., M.L.G., D.F.G., L.J.H., A.H., J.H., R.D.J., D.K., J.K., E.M.L., L.A.L., C.L., X.L., J.L., R.M., A.C.M., S.P., A.P., O.P., D.P., A.P.R., F.R., I.R., C.F.S., D.S., R.A.S., D.St., J.A.V., U.V., D.V., J.G.W and M.Z; study PI: E.B., J.E.B., L.C., D.F.E., C.Ha., F.B.H., S.L., A.M., C.E.P., P.M.R., K.S., E.A.S., D.T., A.G.U., S.U., H.V., N.J.W., M.W., N.F., D.I.C., U.T., A.Mu., Ka.S., J.M.M. and K.K.O.; working group/project management: K.L.L., F.R.D., P.S., K.S.R., T.E., C.E.E., N.F., D.I.C., U.T., A.Mu., Ka.S., J.M.M., K.K.O and J.R.B.P.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-5 and Supplementary Tables 1-3 (PDF 1927 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lunetta, K., Day, F., Sulem, P. et al. Rare coding variants and X-linked loci associated with age at menarche. Nat Commun 6, 7756 (2015). https://doi.org/10.1038/ncomms8756
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8756
This article is cited by
-
Polygenic interactions with adiposity rebound in the prediction of thelarche
Pediatric Research (2021)
-
Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity
Nature Genetics (2018)
-
Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood
Scientific Reports (2016)
-
Molecular insights into the aetiology of female reproductive ageing
Nature Reviews Endocrinology (2015)
-
Shared genetic aetiology of puberty timing between sexes and with health-related outcomes
Nature Communications (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
