
Abstract
Malignant mixed Müllerian tumours, also known as carcinosarcomas, are rare tumours of gynaecological origin. Here we perform whole-exome analyses of 22 tumours using massively parallel sequencing to determine the mutational landscape of this tumour type. On average, we identify 43 mutations per tumour, excluding four cases with a mutator phenotype that harboured inactivating mutations in mismatch repair genes. In addition to mutations in TP53 and KRAS, we identify genetic alterations in chromatin remodelling genes, ARID1A and ARID1B, in histone methyltransferase MLL3, in histone deacetylase modifier SPOP and in chromatin assembly factor BAZ1A, in nearly two thirds of cases. Alterations in genes with potential clinical utility are observed in more than three quarters of the cases and included members of the PI3-kinase and homologous DNA repair pathways. These findings highlight the importance of the dysregulation of chromatin remodelling in carcinosarcoma tumorigenesis and suggest new avenues for personalized therapy.
Similar content being viewed by others


Introduction
Malignant mixed Müllerian tumours (MMMT) are infrequent gynaecological cancers with histological features of both carcinoma and sarcoma1. These tumours arise most commonly in the uterus2, but have also been identified in the vagina3, cervix4, ovary5, fallopian tube6 and peritoneum7. Despite affecting only two per 100,000 US women per year, these tumours are highly aggressive and 5-year survival rates have been reported between 35% and 65% in early-stage carcinosarcoma and only around 10% for stage IV neoplasms8.
MMMT typically affect women over the age of 50 years, with a median age of onset of 62 years9. Patients may present with vaginal bleeding, abdominal pain and/or mass, but are generally not diagnosed until the disease is in late stage10. In addition to advanced age, risk factors include nulliparity, obesity, exogenous oestrogen exposure, pelvic irradiation and treatment with Tamoxifen9,11,12. The primary treatment for MMMT is total abdominal hysterectomy with bilateral salpingo-oophorectomy13. In addition to surgery, high rates of recurrence and metastasis necessitate the use of adjuvant chemotherapy and radiation13. Despite these interventions, the prognosis is poor and more effective therapeutic options are needed to aid the clinical management of affected patients.
There are three theories for the origin of carcinosarcomas. The collision theory suggests that the tumours are biclonal arising from separate cells that later merge. The combination theory assumes that a common precursor differentiates bidirectionally and the conversion theory posits that a single cell undergoes metaplasmic differentiation9,10. Molecular and histological evidence supports the conversion hypothesis14 and these tumours are now thought to derive from sarcomatous differentiation in a high-grade carcinoma10.
Alterations in TP53, KRAS and the PIK3CA pathway have previously been observed in 25%, 15% and 19% of carcinosarcomas, respectively15,16. Mutations of CTNNB1 and NRAS, as well as mismatch repair deficiency, are known to occur at a lower frequency15,16,17. However, the genetic basis of this tumour type remains largely unexplored. We report here the results of the first unbiased systematic genetic analysis of carcinosarcomas using whole-exome sequencing. These analyses define the mutational landscape of carcinosarcoma and reveal the importance of chromatin remodelling in this tumour type.
Results
Overall approach and spectrum of somatic mutations
We used a next-generation sequencing approach to analyse the entire exomes (>20,000 genes) of matched tumour and normal specimens from 22 carcinosarcoma patients for tumour-specific (somatic) mutations. All samples were primary tumours, with the exception of MM02, which was a recurrence at the primary site, and included 17 tumours originating in the uterus and 5 tumours arising in the ovaries (Supplementary Table 1). On average, the sequencing coverage was 191-fold and >90% of bases were covered by at least 10 high-quality reads (Supplementary Table 2). The number of somatic non-synonymous mutations in 18 of these tumours ranged from 4 to 88, with an average of 43 mutations per tumour. The number of somatic mutations per tumour was similar to that of other gynaecological cancers, including ovarian clear cell18, high-grade ovarian serous19 and uterine serous20 carcinomas, but significantly higher than previously observed in low-grade ovarian cancers and pediatric tumours21,22,23. Four tumours harboured a significantly higher number of somatic alterations, ranging from 904 to 5,913. Further analyses revealed somatic mutations in the mismatch repair genes, MLH1 (MM20T) and MSH6 (MM04T, MM12T and MM18T) in all cases, suggesting that these defects were responsible for the observed mutator phenotype (Supplementary Table 3). MM12T harboured truncating mutations in MSH6 in both alleles, whereas single nonsense or frameshift mutations were observed in the other three cases. Microsatellite instability has been previously reported in type I endometrial cancers and low-grade ovarian tumours, as well as in carcinosarcomas16,17,21,24.
The vast majority of somatic alterations observed were single base substitutions, including non-synonymous coding changes, nonsense mutations and splice site alterations, with the remaining mutations as small insertions and deletions (Supplementary Table 3). The non-silent substitutions in the microsatellite stable cases were predominantly C:G to T:A transitions (Fig. 1). These mutations are thought to arise as a result of deamination of 5-methyl cytosine at CpG sites25 and are a common feature across cancer types, including other gynaecological tumours18,26. Mismatch repair-defective cases displayed an atypical mutational signature consistent with previous reports26,27,28. Almost half of the somatic mutations in the tumour harbouring an MLH1 alteration were small insertions and deletions, compared with only 10% in the microsatellite stable cases. In the MSH6-mutated cases, C:G to T:A transitions were the most common, accounting for 53% of somatic mutations, but there was also an overrepresentation of C:G to A:T transversions (30%), probably as a consequence of the inability to repair mismatches arising as a result of oxidative damage28 (Fig. 1).

(a) The frequency of non-synonymous mutations in each sample of the study is displayed as mutations per megabase. (b) The matrix represents individual mutations in 22 patient samples, colour coded by type of mutation. Only the most damaging mutation per gene is shown if multiple mutations were found in a sample. Genes are classified into four functional categories displayed on the right. Mutations occurring at known hotspots are circled in green. Left: bar plot shows the number of mutations in each gene across 22 samples. Percentages represent the fraction of tumours with at least one mutation in the specified gene. (c) Base substitution spectrum of individual samples: transitions (ts) and transversions (tv) at CpG and non-CpG sites, indels and dinucleotide substitutions. (d) Site of the primary tumour.
Recurrently mutated genes
Overall, we identified 777 somatic mutations in 702 genes in the 18 microsatellite stable cancers, as well as 18,371 alterations in 4 mismatch repair-deficient tumours. The analyses of tumour-specific mutations revealed a number of known genes with recurrent alterations. Mutations in TP53 were observed in 67% (16/22) of cases, a significantly higher fraction than previously appreciated15. The phosphoinositide 3-kinase (PI3-kinase) pathway was affected by activating mutations in PIK3CA (9/22 cases) and PTEN (9/22 cases). We identified mutations in PIK3R1, a member of the PI3-kinase pathway that had not been previously implicated in carcinosarcomas, in 3 of 22 cases. As in endometrial tumours24, mutations in PIK3CA and PIK3R1 were mutually exclusive, but loss of PTEN function co-occurred with other genes in this pathway. Unlike in previous studies of carcinosarcomas, PI3-kinase pathway mutations were not confined to tumours located in the uterus, but also occurred in ovarian cases15. Hotspot mutations in CTNNB1 were observed in two microsatellite stable cases (Fig. 1).
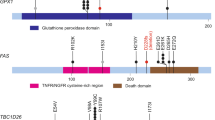
In addition to genes already associated with carcinosarcomas, we identified mutations in genes not previously implicated in this tumour type (Fig. 1). Truncating mutations in ARID1A were detected in 32% (7/22) of tumours (Figs 1 and 2). Four of the five ovarian tumours had ARID1A mutations and these were not observed in the microsatellite-stable tumours of the uterus. Three of the ARID1A-mutated tumours also harboured frameshift and nonsense alterations in ARID1B. Alterations in ARID1A have been reported in other gynaecological cancers, including a high proportion of ovarian clear cell carcinoma (57%)18, endometrioid carcinoma of the ovary29 and endometrial tumours24, as well as a range of other tumour types such as neuroblastoma, colorectal, gastric, prostate and breast cancers23,30. Intragenic deletions and point mutations in ARID1B were recently discovered in around 10% of neuroblastomas23 and have been seen in a small fraction of hepatocellular31, breast32 and medulloblastoma tumours33,34.

Locations of mutations in the ARID1A, ARID1B, SPOP, BAZ1A and MLL3 proteins are indicated with arrows. Red arrows indicate mutations predicted to truncate the protein, while black arrows indicate missense alterations. Each tumour sample is depicted in a different colour.
Alterations of the histone methyltransferase MLL3 were identified in 27% (6/22) of carcinosarcomas. One of these tumours also harboured a splice site alteration in the related gene, MLL2. Mutations in these genes have been observed in ovarian clear cell carcinomas18 and are known to be involved in the tumorigenesis of medulloblastoma22, lung squamous cell carcinoma35 and Mantle cell lymphoma36. In addition, SPOP, a putative tumour suppressor involved in chromatin remodelling was found to be altered in 3 cases (14%). Mutations in SPOP have been previously reported in endometrial37, prostate38, lung39 and colorectal cancers40. These mutations fall nearby the reported alterations within the MATH domain and the encoded protein is known to modulate the transcriptional repression activities of DAXX, a component of the histone deacetylase co-repressor complex41. BAZ1A, an accessory unit of the ATP-dependent chromatin assembly factor42 was mutated in 4 of the 22 carcinosarcomas. BAZ1A is mutated at a low frequency in ovarian cancer19 and deletions of this gene are known to occur in renal papillary carcinoma43. Overall, approximately two-thirds of cases harboured mutations in genes involved in chromatin remodelling or modification.
Mutations in the tumour suppressor gene, FBXW7, were identified in 23% (5/22) of cases. The encoded protein is a member of the SCF (SKP-cullin-F-box) ubiquitin ligase complex that targets cyclin E for degradation44. One mutation was a frameshift predicted to truncate the protein and the other three missense alterations were clustered in the WD repeat domain. Tumour MM19 carried the previously described hotspot mutation, 505R>C. FBXW7 is mutated in a number of different tumour types, including colorectal and haematopoietic malignancies, and has been recently reported in approximately a third of endometrial cancers20,37.
The serine/threonine phosphatase, PPP2R1A, was mutated in one uterine carcinosarcoma. Alterations in this gene were initially identified in ovarian clear cell carcinomas18 and more recently in a range of tumour types, including a number of endometrial cancers and across different ovarian cancer subtypes24,45. The missense mutation, 180M>R, falls nearby other hotspot mutations within the HEAT domain18,24.
Consistent with the accepted theory of carcinosarcomas arising from a single cell, mutations in known cancer genes identified through whole-exome analyses appeared to be clonal in nature. Seven of the cases had TP53 mutation frequencies above 80%, indicating a homozygous change in all tumour cells when adjusted for the presence of contaminating normal tissue, while heterozygous mutations in TP53 or other known cancer genes in 10 additional cases were present at an average of 49.5%. These analyses suggest that individual carcinosarcomas comprises a population of cells that contain largely identical somatic sequence alterations.
Clinically actionable alterations
There are currently no targeted therapies for carcinosarcomas and the prognosis for patients with this tumour type is poor. We investigated whether the observed mutations may be clinically actionable using existing or investigational therapies. We examined genetic alterations that were associated with Food and Drug Administration-approved therapies for oncologic indications, therapies in published prospective or retrospective clinical studies and ongoing clinical trials for cancer patients. We also evaluated alterations in 84 well-known cancer predisposing genes in the patients’ germline that may affect cancer susceptibility as detection of such changes have important implications for early detection and intervention. We identified mutations in a number of potentially clinically actionable genes (Table 1). The PI3-kinase pathway was activated in over half of the carcinosarcomas through alterations in PIK3CA, PIK3R1 and PTEN. Targeted inhibition of this pathway using PI3K/mammalian target of rapamycin/AKT/MEK inhibitors has been reported and is being studied in ongoing Phase I and II trials in endometrial cancer and other tumour types46,47,48,49. A known hotspot missense mutation, 104V>M, in the extracellular domain of the kinase ERBB3 (ref. 50) was observed in a single tumour, and antibodies targeting this protein have been in clinical development, including phase II studies. Similarly, KRAS hotspot mutations were identified in five cases and tumours of these patients may respond to MEK/BRAF inhibitors. Pre-clinical evidence suggests that loss of FBXW7 function, as observed in a number of the carcinosarcomas, may result in sensitivity to HDAC inhibitors51. Two somatic nonsense mutations in BRCA2, a frameshift in BRCA1 and a splice site alteration of FANCM were also identified providing further evidence for the role of homologous recombination repair defects in carcinosarcomas52. In addition, we identified a nonsense mutation (3326K>X) in the BRCA2 gene in the germline of a patient (MM08) with a family history of cervical (mother) and lung cancer (sister). Tumours of these patients may be sensitive to DNA cross-linking agents and to poly (ADP-ribose) polymerase inhibitors53,54. The relatively high frequency of mismatch repair-deficient tumours may also be clinically significant, as the efficacy of the antibody, MK-34775, is actively being evaluated in patients with microsatellite unstable tumours.
Discussion
These analyses have provided the most comprehensive characterization of the mutational landscape of carcinosarcomas to date. Our data show that carcinosarcomas have one of the highest frequencies of chromatin remodelling dysregulation of all tumour types analysed to date. The most highly mutated genes, ARID1A, along with ARID1B, are key components of the conserved, ATP-dependent SWI/SNF chromatin remodelling complex55. This complex uses helicase activity to allow transcription factors access to DNA56 and is important in the regulation of multiple cellular processes, including DNA repair, cell cycle progression and cell migration57,58. Inactivation of the ARID1 complex appears to be particularly important in gynaecological cancers as observed in this study and in other tumours of the female genital tract18,24,29. Among the microsatellite-stable tumours, ARID1A mutations were unique to carcinosarcomas originating in the ovaries, which may suggest different mutational signatures in carcinosarcomas of different sites. This is a potentially significant finding as the prognosis for ovarian carcinosarcomas tends to be better than for those of the uterus8.
Other genes involved in chromatin modification in this tumour type include MLL3, SPOP and BAZ1A. MLL3 encodes a histone H3K4 trimethylase that is part of the ASC-2 complex (ASCOM). This complex is a co-activator of TP53 and regulates the expression of TP53 target genes in response to DNA damage59. Members of the MLL family have also been shown to have a role in HOX gene expression60 and Wnt signalling61. SPOP is a BTB (Bric-a-brac/Tramtrack/Broad complex) protein that recruits DAXX to the ubiquitin ligase Cul3, resulting in ubiquitination and degradation of the DAXX protein62. This leads to the reversal of DAXX-mediated repression of ETS1 and p53-dependent transcription62. The BAZ1A gene encodes the accessory subunit of the ATP-dependent chromatin assembly factor. This complex generates and maintains nucleosome spacing, a function critical for chromatin condensation and appropriate gene silencing42. These observations suggest that a variety of different aspects of chromatin regulation and modification are genetically altered in carcinosarcomas. Genomic analyses in other tumour types have shown that somatic mutations of epigenetic regulators can have important clinical consequences, including improved outcome in patients with DAXX/ATRX alterations in pancreatic neuroendocrine tumours63 and gliomas64, and a decreased survival in patients with ARID1A and ARID1B mutations in neuroblastoma23.
The presence of carcinoma and sarcomatous components in MMMTs has hindered the clinical management of patients, as the prognosis and appropriate therapies for carcinomas and sarcomas differ. These molecular data support the idea that carcinosarcomas are most similar to carcinomas, based on the presence of alterations in genes such as PTEN that have only rarely been observed in endometrial sarcomas65 and uterine sarcomas66, but are common in uterine adenocarcinomas67. In addition, the number of non-synonymous mutations observed in the carcinosarcomas was similar to that reported in carcinomas and significantly higher than is typical for sarcomas analysed to date19,68. Our genomic analyses suggest that the unusual pathology of this tumour type could be related to the loss of control of genetic programing as a result of mutations in chromatin remodelling genes in the specific context of progenitor cells that lead to carcinosarcomas.
In addition to providing therapeutic insight, understanding the mutational profile of MMMTs will be beneficial for diagnostic purposes, including early detection, which can immediately have an impact on survival. Recent studies have used mutations as highly specific biomarkers heralding the presence of malignancy even at the earliest stages by sequencing cell-free DNA in the circulation as well as DNA extracted from liquid Papanicolaou smear specimens for the detection of gynaecological cancers69,70.
Defining the mutational landscape of carcinosarcomas has highlighted alterations in specific genes and pathways that may aid the diagnosis and future clinical management of patients with this malignancy. Alterations of the PI3K and DNA repair pathways have identified specific actionable targets, which have not been previously considered in this tumour type, as well as a frequent dysregulation of chromatin remodelling. These analyses suggest future efforts at defining the downstream targets of chromatin regulators in carcinosarcomas as well as interventional clinical trials based on potentially actionable alterations observed in these cancer patients.
Methods
Samples
Twenty-two carcinosarcoma tumour samples and matched normal tissues were obtained with consent from all study participants and approved for research use by the OHSU Institutional Review Board and the Knight BioLibrary. Eleven patients were treated with chemotherapy or radiation after surgery. Samples underwent pathological review to determine tumour cellularity. Tumours were macrodissected to remove contaminating normal tissue. An average of 85% neoplastic cellularity was obtained. DNA was extracted from 10 fresh-frozen tumours, 12 formalin-fixed paraffin embedded tumours and matched normal tissue samples (Supplementary Table 1).
Library generation and sequencing
Genomic DNA from tumour and normal samples were fragmented and used for Illumina TruSeq library construction (Illumina, San Diego, CA). Exonic regions were captured in solution using the Agilent SureSelect 51 Mb kit (version 4) according to the manufacturer’s instructions (Agilent, Santa Clara, CA). Paired-end sequencing, resulting in 100 bases from each end of the fragments, was performed using a HiSeq 2000 Genome Analyzer (Illumina).
Bioinformatic analyses
Sequencing reads were analysed and aligned to human genome hg19 with the Eland algorithm in CASAVA 1.7 software (Illumina). Reads were mapped using the default seed-and-extend algorithm, which allowed a maximum of two mismatched bases in the first 32 bp of sequence. Identification of somatic alterations was performed using a next-generation sequencing analysis pipeline that enriched for tumour-specific single-nucleotide alterations and small indels. Briefly, for each position with a mismatch (compared with the hg19 reference sequence using the Eland algorithm), the read coverage of the mismatched and wild-type sequence at that base was calculated. For determination of tumour-specific alterations, tumour and matched normal sequences were compared and known polymorphisms were removed from the analysis. Potential somatic mutations were filtered and visually inspected, and a candidate-mismatched base was identified as a mutation when distinct paired tags contained the mismatched base and the mismatched base was not present in the matched normal sample.
Additional information
Accession codes: Carcinosarcoma exome sequence data has been deposited at the European Genome-phenome Archive ( http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under the accession code EGAS00001000941.
How to cite this article: Jones, S. et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat. Commun. 5:5006 doi: 10.1038/ncomms6006 (2014).
References
Jin, Z. et al. Carcinosarcomas (malignant mullerian mixed tumors) of the uterus and ovary: a genetic study with special reference to histogenesis. Int. J. Gynecol. Pathol. 22, 368–373 (2003).
El-Nashar, S. A. & Mariani, A. Uterine carcinosarcoma. Clin. Obstet. Gynecol. 54, 292–304 (2011).
Ahuja, A., Safaya, R., Prakash, G., Kumar, L. & Shukla, N. K. Primary mixed mullerian tumor of the vagina--a case report with review of the literature. Pathol. Res. Pract. 207, 253–255 (2011).
Sharma, N. K., Sorosky, J. I., Bender, D., Fletcher, M. S. & Sood, A. K. Malignant mixed mullerian tumor (MMMT) of the cervix. Gynecol. Oncol. 97, 442–445 (2005).
Duman, B. B., Kara, I. O., Gunaldi, M. & Ercolak, V. Malignant mixed Mullerian tumor of the ovary with two cases and review of the literature. Arch. Gynecol. Obstet. 283, 1363–1368 (2011).
Shen, Y. M. et al. Malignant mixed mullerian tumor of the fallopian tube: report of two cases and review of literature. Arch. Gynecol. Obstet. 281, 1023–1028 (2010).
Rajanbabu, A., Zahoor, A. S., Vijajkumar, D. K., Pavithran, K. & Kuriakose, S. The significance of the site of origin in primary peritoneal carcinosarcoma: case report and literature review. Ecancermedicalscience 7, 295 (2013).
George, E. M. et al. Carcinosarcoma of the ovary: natural history, patterns of treatment, and outcome. Gynecol. Oncol. 131, 42–45 (2013).
Kanthan, R. & Senger, J. L. Uterine carcinosarcomas (malignant mixed mullerian tumours): a review with special emphasis on the controversies in management. Obstet. Gynecol. Int. 2011, 470795 (2011).
Kernochan, L. E. & Garcia, R. L. Carcinosarcomas (malignant mixed Mullerian tumor) of the uterus: advances in elucidation of biologic and clinical characteristics. J. Natl Compr. Canc. Netw. 7, 550–556 quiz 557 (2009).
Meredith, R. F. et al. An excess of uterine sarcomas after pelvic irradiation. Cancer 58, 2003–2007 (1986).
Curtis, R. E., Freedman, D. M., Sherman, M. E. & Fraumeni, J. F. Jr Risk of malignant mixed mullerian tumors after tamoxifen therapy for breast cancer. J. Natl Cancer Inst. 96, 70–74 (2004).
Callister, M., Ramondetta, L. M., Jhingran, A., Burke, T. W. & Eifel, P. J. Malignant mixed Mullerian tumors of the uterus: analysis of patterns of failure, prognostic factors, and treatment outcome. Int. J. Radiat. Oncol. Biol. Phys. 58, 786–796 (2004).
Armstrong, A. B. et al. TP53 mutational analysis supports monoclonal origin of biphasic sarcomatoid urothelial carcinoma (carcinosarcoma) of the urinary bladder. Mod. Pathol. 22, 113–118 (2009).
Growdon, W. B. et al. Tissue-specific signatures of activating PIK3CA and RAS mutations in carcinosarcomas of gynecologic origin. Gynecol. Oncol. 121, 212–217 (2011).
de Jong, R. A. et al. Molecular markers and clinical behavior of uterine carcinosarcomas: focus on the epithelial tumor component. Mod. Pathol. 24, 1368–1379 (2011).
South, S. A., Hutton, M., Farrell, C., Mhawech-Fauceglia, P. & Rodabaugh, K. J. Uterine carcinosarcoma associated with hereditary nonpolyposis colorectal cancer. Obstet. Gynecol. 110, 543–545 (2007).
Jones, S. et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231 (2010).
Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Kuhn, E. et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J. Natl Cancer Inst. 104, 1503–1513 (2012).
Jones, S. et al. Low-grade serous carcinomas of the ovary contain very few point mutations. J. Pathol. 226, 413–420 (2012).
Parsons, D. W. et al. The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439 (2011).
Sausen, M. et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 45, 12–17 (2013).
Cancer Genome Atlas Research Network. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
Pfeifer, G. P. Mutagenesis at methylated CpG sequences. Curr. Top. Microbiol. Immunol. 301, 259–281 (2006).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Pena-Diaz, J. et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. Cell 47, 669–680 (2012).
Ni, T. T., Marsischky, G. T. & Kolodner, R. D. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell 4, 439–444 (1999).
Wiegand, K. C. et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 363, 1532–1543 (2010).
Jones, S. et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 33, 100–103 (2012).
Fujimoto, A. et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 44, 760–764 (2012).
Stephens, P. J. et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404 (2012).
Jones, D. T. et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105 (2012).
Pugh, T. J. et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110 (2012).
Kim, Y. et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in east asian patients. J. Clin. Oncol. 32, 121–128 (2013).
Bea, S. et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl Acad. Sci. USA 110, 18250–18255 (2013).
Le Gallo, M. et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 44, 1310–1315 (2012).
Berger, M. F. et al. The genomic complexity of primary human prostate cancer. Nature 470, 214–220 (2011).
Kan, Z. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 (2010).
Kim, M. S., Je, E. M., Oh, J. E., Yoo, N. J. & Lee, S. H. Mutational and expressional analyses of SPOP, a candidate tumor suppressor gene, in prostate, gastric and colorectal cancers. APMIS 121, 626–633 (2013).
Hollenbach, A. D., McPherson, C. J., Mientjes, E. J., Iyengar, R. & Grosveld, G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell. Sci. 115, 3319–3330 (2002).
Racki, L. R. et al. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature 462, 1016–1021 (2009).
Krill-Burger, J. M. et al. Renal cell neoplasms contain shared tumor type-specific copy number variations. Am. J. Pathol. 180, 2427–2439 (2012).
Welcker, M. & Clurman, B. E. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93 (2008).
Rahman, M. et al. PPP2R1A mutation is a rare event in ovarian carcinoma across histological subtypes. Anticancer Res. 33, 113–118 (2013).
Moroney, J. et al. Phase I study of the antiangiogenic antibody bevacizumab and the mTOR/hypoxia-inducible factor inhibitor temsirolimus combined with liposomal doxorubicin: tolerance and biological activity. Clin. Cancer Res. 18, 5796–5805 (2012).
Fu, S. et al. Perifosine plus docetaxel in patients with platinum and taxane resistant or refractory high-grade epithelial ovarian cancer. Gynecol. Oncol. 126, 47–53 (2012).
Seront, E. et al. Phase II study of everolimus in patients with locally advanced or metastatic transitional cell carcinoma of the urothelial tract: clinical activity, molecular response, and biomarkers. Ann. Oncol. 23, 2663–2670 (2012).
Slomovitz, B. M. et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 116, 5415–5419 (2010).
Jaiswal, B. S. et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617 (2013).
Garnett, M. J. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575 (2012).
Pennington, K. P. et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 20, 764–775 (2013).
Villarroel, M. C. et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 10, 3–8 (2011).
Williamson, C. T. et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 4, 515–527 (2012).
Wang, X. et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem. J. 383, 319–325 (2004).
Wilson, B. G. & Roberts, C. W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492 (2011).
Weissman, B. & Knudsen, K. E. Hijacking the chromatin remodeling machinery: impact of SWI/SNF perturbations in cancer. Cancer Res. 69, 8223–8230 (2009).
Caramel, J., Quignon, F. & Delattre, O. RhoA-dependent regulation of cell migration by the tumor suppressor hSNF5/INI1. Cancer Res. 68, 6154–6161 (2008).
Lee, J. et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl Acad. Sci. USA 106, 8513–8518 (2009).
Ansari, K. I. & Mandal, S. S. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J. 277, 1790–1804 (2010).
Sierra, J., Yoshida, T., Joazeiro, C. A. & Jones, K. A. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 20, 586–600 (2006).
Kwon, J. E. et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J. Biol. Chem. 281, 12664–12672 (2006).
Jiao, Y. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–1203 (2011).
Jiao, Y. et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3, 709–722 (2012).
Amant, F. et al. PTEN mutations in uterine sarcomas. Gynecol. Oncol. 85, 165–169 (2002).
Lancaster, J. M., Risinger, J. I., Carney, M. E., Barrett, J. C. & Berchuck, A. Mutational analysis of the PTEN gene in human uterine sarcomas. Am. J. Obstet. Gynecol. 184, 1051–1053 (2001).
Risinger, J. I., Hayes, A. K., Berchuck, A. & Barrett, J. C. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res. 57, 4736–4738 (1997).
Barretina, J. et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 42, 715–721 (2010).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra24 (2014).
Haber, D. A. et al. Blood-Based Analyses of Cancer: Circulating Tumor Cells and Circulating Tumor DNA. Cancer Discov 4, 650–661 (2014).
Acknowledgements
We thank David R. Riley and Karli Lytle for their assistance with next-generation sequencing analyses. This work was supported in part by Dr Miriam and Sheldon G. Adelson Medical Research Foundation, the Commonwealth Foundation, Swim Across America, and NIH grant CA121113.
Author information
Authors and Affiliations
Contributions
S.J. and N.S. performed the analyses, and wrote and revised the paper. C.L.M., E.C., S.V.A. and M.S. analysed the data. J.L. and D.K. collected the specimens and provided clinical information. L.K. and M.S. carried out the experimental work. R.M. and L.D.W. provided histopathological confirmation. L.A.D., C.L. and V.E.V. conceived the study, and wrote and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
L.A.D. and V.E.V. are co-founders of Personal Genome Diagnostics and are members of its Scientific Advisory Board and Board of Directors. L.A.D. and V.E.V. own Personal Genome Diagnostics stock, which is subject to certain restrictions under University policy. The terms of these arrangements are managed by the Johns Hopkins University in accordance with its conflict-of-interest policies. The remaining authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Tables 1-3 (PDF 16048 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jones, S., Stransky, N., McCord, C. et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 5, 5006 (2014). https://doi.org/10.1038/ncomms6006
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6006
This article is cited by
-
Leveraging the replication stress response to optimize cancer therapy
Nature Reviews Cancer (2023)
-
Clinical significance of SPOP and APC gene alterations in colorectal cancer in Indian population
Molecular Genetics and Genomics (2023)
-
Genomic alterations in gynecological malignancies: histotype-associated driver mutations, molecular subtyping schemes, and tumorigenic mechanisms
Journal of Human Genetics (2021)
-
Clinical actionability of molecular targets in endometrial cancer
Nature Reviews Cancer (2019)
-
Sarcomatoid carcinomas of the gallbladder: clinicopathologic characteristics
Virchows Archiv (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
