
Abstract
Field-effect transistor biomolecular sensors based on low-dimensional nanomaterials boast sensitivity, label-free operation and chip-scale construction. Chemical vapour deposition graphene is especially well suited for multiplexed electronic DNA array applications, since its large two-dimensional morphology readily lends itself to top-down fabrication of transistor arrays. Nonetheless, graphene field-effect transistor DNA sensors have been studied mainly at single-device level. Here we create, from chemical vapour deposition graphene, field-effect transistor arrays with two features representing steps towards multiplexed DNA arrays. First, a robust array yield—seven out of eight transistors—is achieved with a 100-fM sensitivity, on par with optical DNA microarrays and at least 10 times higher than prior chemical vapour deposition graphene transistor DNA sensors. Second, each graphene acts as an electrophoretic electrode for site-specific probe DNA immobilization, and performs subsequent site-specific detection of target DNA as a field-effect transistor. The use of graphene as both electrode and transistor suggests a path towards all-electrical multiplexed graphene DNA arrays.
Similar content being viewed by others

Introduction
Over the past couple of decades, electrical detection of biomolecules via their inherent charges has undergone an intense renaissance, with the advent of low-dimensional nanomaterials, such as semiconductor nanowires, carbon nanotubes, and more recently, graphene1,2,3,4,5,6,7,8. Configured as field-effect transistors (FETs), these nanomaterials can achieve electrical detection sensitivity down to the picomolar/femtomolar (pM/fM) and even sub-fM level2,4,7,8,9,10. Given the sensitivity, combined with such advantages as label-free operation and bona fide chip-scale construct, nanomaterial-based electrical biomolecular sensors may one day become an important practical tool in biotechnology.
Among the various nanomaterials capable of electrical biomolecular sensing, graphene is particularly well suited for parallel, multiplexed applications11,12, such as DNA microarrays (for which the currently dominant tool resorts to optical methods13,14,15). This is because the large-area planar morphology of graphene—if, for example, grown by chemical vapour deposition (CVD)—is amenable to top-down fabrication for defining a large array of FET sensor sites7,12,16. In fact, graphene FET sensor arrays have been developed for pH monitoring and chemical sensing6. By contrast, studies of graphene FET DNA sensors have so far been focused mainly on single devices17,18,19. To advance a step towards multiplexed graphene DNA arrays, there are critical needs to demonstrate a high-yield array with each FET sensor exhibiting a high sensitivity. In addition, investigation of electrical methods to site-specifically immobilize probe DNA—as an alternative to the lithographic assembly of probe DNA arrays13,15—may prove beneficial.
Here we create an 8-FET DNA sensor array fabricated from CVD graphene, with two key features representing steps towards multiplexed graphene DNA arrays. First, 7 out of the 8 FETs achieve a maximum sensitivity of 100 fM, demonstrating a robust array yield with the sub-picomolar sensitivity. This sensitivity is 10 times higher than the prior state-of-the-art CVD graphene FET DNA sensor18 and is on par with—if not better—the ~1 pM sensitivity of industry-standard optical DNA microarrays14,15,20, which finds a range of molecular diagnostic applications in research and clinical settings. While certain high-end optical and electrochemical DNA sensors21,22,23,24,25,26,27,28 achieve a far higher sensitivity down to the sub-fM level, our device is more advantageous in achieving chip-scale integration and label-free operation simulataneously4,9,18. Second, each graphene site in the array is used not only as a sensing FET but also as an electrophoretic electrode to enable site-specific immobilization of probe DNA. Specifically, when probe DNA of a particular sequence desired at specific graphene sites is introduced into the electrolyte overlying the device array, these graphene sites are positively biased to attract and anchor the probe DNA molecules, while the rest of the graphene sites are negatively biased to repel them. This use of graphene as an electrophoretic electrode for DNA immobilization is an adaptation of electrophoretic DNA immobilization using conducting electrodes of different materials (for example, Pt, Au and carbon paste)29,30,31,32. The resulting site-specific single-stranded probe DNA array then enables site-specific detection of single-stranded target DNA upon hybridization, where this detection phase uses each graphene as a FET. That is, graphene engaged in the dual role of electrophoretic electrode and sensing FET enables site-specific operation of graphene DNA array in an all-electrical manner (in both probe assembly and target detection), offering possibilities for highly multiplexed graphene DNA arrays.
Results
Fabrication of 8-FET graphene array
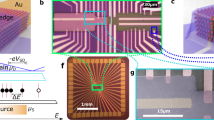
The array fabrication starts by transferring CVD-grown, monolayer-dominated graphene onto a SiO2/Si substrate (Fig. 1; Supplementary Figs 1 and 2)33,34. We pattern this graphene sheet into a linear array of eight local graphene sites by oxygen-plasma etching, with a graphene-to-graphene pitch of 240 μm. Contacts are made to both ends—‘source’ and ‘drain’—of each graphene site with Cr/Au/Cr metals, with the remaining graphene channel being 45-μm long and 90-μm wide (Fig. 1). To obviate the leakage current from these metal contacts to the electrolyte35,36, we passivate the metal contacts by depositing a SiO2 layer. A polydimethylsiloxane microfluidic channel is fabricated on top of the array to facilitate robust, controllable delivery and clearing of DNA molecules as well as materials to functionalize graphene surface. An Ag/AgCl wire immersed in the electrolyte—inserted into the outlet tube—serves as a reference electrode.

(a) Illustration and image of an 8-graphene-electrode/FET array with a microfluidic channel on top. This entire device sits on a printed circuit board. (b) Cross-sectional illustration of an individual graphene site. (c) Optical micrograph of a portion of a fabricated graphene array. For an individual graphene site, W=90 μm and L=45 μm (scale bar, 120 μm). (d) Raman spectrum of a graphene sample transferred onto a SiO2/Si substrate and integrated graphene Raman two-dimensional (2D) peak intensity map from 2,600 cm−1 to 2,800 cm−1 (scale bar, 1 μm).
Passive probe DNA immobilization and target DNA detection
Before the site-specific operation of the array with each graphene site acting as an electrophoretic electrode as well as a sensing FET, we first passively immobilize probe DNA molecules of the same sequence all across the array without site specificity—that is, here graphene is not used as an electrode for site-specific immobilization—and measure the sensitivity limit of each graphene FET. This particular experiment, where each site represents the repetition of the same probe DNA sequence, is to characterize the array sensitivity yield under the same condition for all sites. For the passive immobilization, the reference electrode voltage, VREF, as well as the voltages of the source and drain of each graphene site, VD and VS, are set at 0 V. Under this bias, we sequentially introduce biotinylated bovine serum albumin (BSA)37, streptavidin and biotinylated 41-mer single-stranded probe DNA (Fig. 2a); the probe DNA molecules have a 200 nM concentration in a 0.9 × phosphate-buffered saline (PBS) solution, and 10 min are allowed for their immobilization. Note that our surface functionalization associated with the probe immobilization adopts the standard biotin–streptavidin-binding strategy on BSA adsorbed onto graphene, where the use of BSA on graphene for biomolecular sensing has been demonstrated8,37. After the immobilization of the same-sequence probe DNA molecules on all graphene sites, we remove itinerant DNA molecules with PBS rinsing. Subsequently, 20-mer single-stranded target DNA molecules, whose sequence is complementary to a 20-base segment of the single-stranded probe DNA (Methods), are introduced in 1 × PBS (100 fM), with 4 h allowed for hybridization. The high (1 × ) PBS concentration during this 4-h hybridization phase is to decrease the Debye length to ~0.76 nm (refs 10, 38), thus to reduce the range of electrostatic repulsion between probe and target DNA molecules, promoting their hybridization. Incidentally, the target DNA sequence is complementary to a 20-base segment of the 22-mer microRNA (miRNA) let7g, whose expression level is closely linked to human cancer39,40 (Methods).

(a) Sequence of steps for BSA–streptavidin surface functionalization (I, II), non-electrophoretic probe DNA immobilization (III) and hybridization or control experiments (IV). (b) Measured VREF−IDS curves (five forward VREF sweeps; device 6) right before and after 100-fM target DNA hybridization. VS=−0.03 V and VD=+0.03 V. (c) V0 values extrapolated from the data of b. ΔV0 is the shift from the average of the pre-hybridization V0 values to that of the post-hybridization V0 values. (d) ΔV0 data for 100 fM hybridization and control for all 8 graphene FETs. Statistics are based on five VREF–IDS curve measurements right before and after the phase IV. Results from both forward and reverse VREF sweeps are shown. ΔV0 averaged across all eight graphene FETs, subsuming both forward and reverse sweeps, is also shown for hybridization and control. For all data here: *P<0.05; **P<0.01; no *, not significant; and the error bars represent ±1 s.d.
Since the inherent negative charges of hybridized target DNA molecules cause extra electron doping of each graphene site17,18,41, hybridization can be detected by monitoring the negative shift of the charge-neutrality point of the graphene site from before to after the 4-h hybridization phase. To measure the charge-neutrality point, we record the source–drain current, IDS, of the individual graphene FET as a function of VREF. To ensure detection confidence in the presence of intrinsic charge-neutrality point fluctuations, for each graphene FET, the VREF–IDS curve is recorded five times—by repeating five times a forward and backward sweep of VREF between 0 and 1 V (throughout this work; each forward or backward sweep lasts ca. 1 min)— both before and after the 4-h hybridization phase. Each individual recording of the VREF–IDS curve is done with a fresh 0.005 × PBS to ensure the same electrochemical condition for each sweep. The low-concentration (0.005 × ) PBS during the VREF–IDS recording increases the Debye length to ~10.7 nm, which enhances the FET sensitivity to the charges of the hybridized target DNA38,42.
Figure 2b displays data from one FET with forward VREF sweeps, that is, five VREF–IDS pre-hybridization-phase curves and five VREF–IDS post-hybridization-phase curves with the 100 fM target DNA. The overall negative shift in the charge-neutrality point, V0, from the pre- to post-hybridization phase is conspicuous, and is distinctively larger than the fluctuations of V0 within the pre- or the post-hybridization phase. This is quantitatively seen in Fig. 2c that displays the V0 values extracted from the data of Fig. 2b (Methods). We define ΔV0 as the shift from the average of the pre-hybridization V0 values to the average of the post-hybridization V0 values. ΔV0 is −27.9 mV, with an appreciably smaller s.d. of ±7.6 mV. Clearly, this graphene FET exhibits a 100-fM sensitivity.
In fact, 7 out of the 8 FETs robustly achieve the 100-fM sensitivity, attesting to the high-yield array performance. This is seen in Fig. 2d, left, where ΔV0 data for all 8 graphene FETs summarize our extensive experiments that entail not only 100 fM hybridization experiments but also 100 fM control experiments, with the control DNA being not complementary to the probe DNA. For the control experiments following the hybridization experiments, we remove hybridized target DNA with a 45-min 90°C deionized water wash, which causes partial loss of probe DNA and surface functionalization, and reintroduce BSA, streptavidin and probe DNA (Supplementary Fig. 3). We measure again VREF–IDS curves (five forward and backward sweeps of VREF) with the probe DNA alone before control DNA is introduced, and measure VREF–IDS curves (five forward and backward sweeps of VREF) 4 h after control DNA is introduced. In 7 FETs, the ΔV0 data of the 100-fM hybridization experiments are significantly separated from those of the control experiments (Fig. 2d, left); only device 4 in Fig. 2d, left, fails to show significant separation in ΔV0 data between the hybridization and control experiments. As shown in Fig. 2d, right, the ΔV0 value averaged over all FETs for the hybridization experiment is −34.5±10.0 mV, while that for the control experiment is only −2.1±5.4 mV, where the average includes even the suboptimal device 4. The control measurement data affirm that the intrinsic drift of the neutrality point during the 4-h elapse is appreciably small—this is ascribable to the large double-layer capacitance at the graphene–electrolyte interface—, and that the distinctive ΔV0 from the hybridization experiments is caused indeed and mainly by the actual hybridization. Finally, juxtaposition of the left and right of Fig. 2d shows that as compared with the overall device average, devices 1, 2, 4, 5, 6 and 7 respond slightly more weakly to hybridization, device 3 slightly more strongly and device 8 far more strongly.
Since graphene is directly doped by the hybridized DNA charges, the graphene FET is inherently more sensitive than the standard bulk ion-sensitive FET whose channel properties are capacitively altered by external charges43,44,45. But beyond this inherent sensitivity, our graphene FET achieves a 10 times higher sensitivity than the prior state-of-the-art CVD graphene FET DNA sensor18. This considerable extra sensitivity is attributed to our opportunistic alteration of the PBS concentration42,45; the high-concentration 1 × PBS during the 4-h hybridization phase reduces the Debye length, promoting hybridization, while the low-concentration 0.005 × PBS during the VREF–IDS recordings increases the Debye length, allowing the FET to feel more of the hybridized DNA charges. In addition, our surface functionalization using the BSA–streptavidin layer shares credit for the increased sensitivity. This negatively charged layer prevents the negatively charged probe DNA molecules from lying flat on the graphene surface, further promoting hybridization38,42. In fact, this may explain, in part, our use of only 200 nM probe DNA molecules—a concentration 5~50 times lower than in other CVD-graphene DNA sensors that do not employ the BSA–streptavidin functionalization17,18—while achieving the sub-picomolar sensitivity.
Site-specific probe DNA immobilization and target detection
We now demonstrate site-specific probe immobilization and target detection (Fig. 3) with a new 8-graphene device array of the same construct as the previous array. To facilitate the proof of concept, we use 200 nM probe DNA molecules of the single identical sequence, but immobilize them only to even-numbered graphene sites (site numbering is in reference to Fig. 3). We achieve this by using each site’s graphene as an electrophoretic electrode, as opposed to a sensing FET, during the probe immobilization phase. Specifically, the even-numbered graphene sites are positively biased with respect to VREF=0 V by setting VS=VD=1.2 V, attracting negatively charged probe DNA molecules. By contrast, odd-numbered sites are negatively biased (VS=VD=−1.2 V), repelling probe DNA molecules. In addition, we apply this electrophoretic site-selection principle not only to the probe DNA molecules in 0.005 × PBS during the immobilization phase (20 s)42, but also to the streptavidin molecules, which are also negatively charged in 0.1 × PBS during the surface functionalization phase (20 s)38 (Fig. 3a). In this way, we further invigorate the site selectivity for the probe DNA immobilization; even-numbered sites have preferentially a far greater number of streptavidin and probe DNA molecules than odd-numbered sites.

(a) During the immobilization of streptavidin and probe DNA, even- (odd-) numbered sites are biased at +1.2 V (−1.2 V) with respect to VREF=0 V for 20 s. (b) Confocal fluorescence image (false coloured) of the array after the site-specific probe DNA immobilization, and the corresponding spatial profile of the averaged fluorescence intensity shown across the array. The averaged fluorescence intensity across the four even-numbered graphene sites is distinctively stronger than that across the four odd-numbered sites. Error bars represent ±1 s.d. *P<0.05. Scale bar, 100 μm. (c) Simulated electrolytic potential profile and spatial pattern of the corresponding electric field magnitude just above the substrate surface, in the setup mimicking the steady-state drift situation (Supplementary Note 4); in the latter, the electric field magnitude of repulsive fields is set to zero and a moving average and normalization to the maximum are applied. (d) Electrical detection of 1 pM hybridization after site-specific DNA immobilization. ΔV0 statistics for each graphene FET are from 5 VREF–IDS measurements before and after 1 pM hybridization, treating forward and reverse VREF sweeps separately. The averaged ΔV0 values subsume both forward and reverse sweeps. Error bars represent ±1 s.d. *P<0.05.
We firmly verify the site-specific immobilization using two distinctive methods, which are confocal fluorescent microscopy and the electrical detection of hybridized target DNAs. The first verification method, confocal fluorescent microscopy, directly maps the spatial profile of immobilized probe DNAs; to this end, we label the probe DNA with fluorophore Cy3. The measured confocal fluorescence image of the graphene array (Fig. 3b) shows the greater concentration of probe DNA molecules in the even-numbered sites and almost no probe DNA molecules in the odd-numbered sites, attesting to the electrophoresis-based site-selective immobilization with a clean-cut finality.
The optical pattern further shows that probe DNA is also immobilized in the non-active regions between adjacent graphene sites, and its density is highest at the non-active regions’ boundaries with even-numbered graphene. This spatial profile can be understood by considering the transient and d.c. fields during the active immobilization process. As we apply the biases as prescribed above, transient electric fields and corresponding transient currents are formed globally across adjacent graphene sites in the bulk of the electrolyte, charging up the double-layer capacitances at the graphene–electrolyte interfaces12,46. This transient process is completed in ca. 30 ms with a relatively large peak current (which is estimated to be ~1.4 μA for 2.4 V; see Fig. 4a and also Supplementary Note 1). As the double-layer capacitances are fully charged, the majority of the applied potentials appear across the nanometre-thick double layers while the bulk of the electrolyte becomes relatively field free46. But even in this steady state, a small d.c. graphene leakage current (which is estimated to be ~1–10 nA at the applied biases; see Fig. 4b and also Supplementary Note 2) still flows globally across adjacent graphene sites, indicating a small steady-state electric field in the electrolyte. These global electric fields across adjacent graphene sites (as opposed to local electric fields within the double layers), both the transient and steady-state d.c., exert a far-reaching electrophoretic force on the probe DNA molecules to attract (repel) them to the positively (negatively) biased graphene sites. This far-reaching force explains the DNA immobilized in the non-active region. Its highest density at the edges of the even-numbered sites is because the global fields are strongest there. The total charge transport, which is proportional to the total probe DNA transport, can be estimated by integrating the current; the d.c. steady-state electric field, although much weaker than the transient peak field, produces more charge transport, because a longer period of ca. 20 s is allotted for the steady-state process (Supplementary Note 3). In fact, the essential pattern of the measured spatial profile of probe DNA density is recapitulated by the simulated global electric field magnitude just above the substrate surface, where the simulation setting mimics the steady-state drift situation (Fig. 3c; Supplementary Note 4).

(a) Measured transient electrolytic current between two neighbouring graphene sites with the application of a voltage step of 1.0 V in 0.005 × PBS. The transient response is complete in ca. 30 ms with a peak current of ~600 nA. From this, we estimate a peak current of ~1.4 μA with a 2.4-V bias difference between two neighbouring graphene sites (Supplementary Note 1). (b) Reference electrode (Ag/AgCl) to graphene voltammogram at a scan rate of 10 mV s−1. From this, the d.c. steady-state leakage current for graphene biased at either +1.2 or −1.2 V is extrapolated to be 1–10 nA (Supplementary Note 2).
In future works, to decrease the deposition of probe DNA in the non-active regions, local integrated reference electrodes can be used, concentrating electric fields near the graphene29,30. Regardless, the key demonstration here is that the electrophoretic procedure immobilizes probe DNA on the positively biased graphene sites, with almost no probe DNA on the negatively biased sites, as confirmed by the optical pattern in agreement with the theoretical consideration.
The second verification of the site-specific probe DNA immobilization is done by site-specific detection of target DNA hybridization (Fig. 3d), for which we engage each graphene site as a sensing FET. After the site-specific immobilization, we introduce 1 pM target DNA molecules in 0.005 × PBS. As expected, the even-numbered graphene FETs where probe DNA molecules have been immobilized exhibit an averaged ΔV0 value of −17.2±5.1 mV with the correct negative shift, whereas the odd-numbered graphene FETs show an appreciably smaller averaged ΔV0 value of -3.1±3.5 mV. In this experiment in Fig. 3, the hybridization detection sensitivity is limited to 1 pM. This is because in the experiment in Fig. 3a, less amount of probe DNA is deposited on the even-numbered graphene sites—as seen by fluorescence image comparison (Supplementary Fig. 3). The amount of immobilized DNA could be enhanced either by allowing for more time for the steady-state process but with a lower bias to ensure that the Faradic current is not damaging or by adopting a.c. electrophoretic biasing such that the majority of the charge transfer is due to non-Faradic currents. Nonetheless, the 1-pM sensitivity is still on a par with standard optical DNA microarrays14,15,20.
Finally, we note that we repeat the same type of site-specific immobilization and detection experiment by reusing the array in Fig. 2 after its surface cleanup. In this additional experiment, the electrical detection results bear the same essence that demonstrates the site-specific immobilization and hybridization; see Supplementary Fig. 4.
Site-specific immobilization of probe DNAs of two sequences
All-electrical site-specific probe immobilization and target detection demonstrated in the foregoing experiment (Fig. 3) is a highlight goal of the present work. The principle may in the future be applied to realizing a fully multiplexed graphene DNA array, where each graphene site would have probe DNA molecules of its own sequence. To assess the technical challenges as well as the feasibility towards this future path, we perform our final experiment with yet another new 8-graphene device array now with probe DNA molecules of two different sequences (Fig. 5), with one sequence labelled with Cy3 fluorophore and the other sequence labelled with Cy5 fluorophore. When the Cy3-labelled probe DNA molecules are introduced in the electrolyte, even-numbered graphene sites are positively biased to attract them, while odd-numbered graphene sites are negatively biased to repel them (Fig. 5a). Subsequently, when the Cy5-labelled probe DNA molecules are introduced, odd-numbered graphene sites are now positively biased to attract them, while even-numbered graphene sites (now immobilized with Cy3-labelled probe DNA molecules) are left unbiased (floating) to prevent possible stripping of the already immobilized probe33. The fluorescence image (Fig. 5b) clearly confirms once again the basic concept of the electrophoresis-based site-specific immobilization, now with the two probe DNA sequences. (We also perform the same type of experiment by once again reusing the array in Fig. 2 after its surface cleanup, and the fluorescence microscopy in this additional experiment once again confirms the site-specific immobilization of the two sequences; see Supplementary Fig. 5.)

(a) During the immobilization of Cy3-labelled probe DNA and associated streptavidin, even- (odd-) numbered sites are biased at +1.2 V (−1.2 V) (20 and 120 s for streptavidin and Cy3-labelled probe DNAs, respectively). During the subsequent immobilization of Cy5-labelled probe DNAs and associated streptavidin, the even- (odd-) numbered sites are left unbiased (biased at +1.2 V), with the same timing scheme as before. (b) Confocal fluorescence image (false coloured) of the array with Cy3 and Cy5 probe DNAs shown, respectively, as red and green, and the corresponding normalized fluorescence intensity. The averaged fluorescence intensities across the four even-numbered and four odd-numbered graphene sites with Cy5 intensity are normalized to the maximum Cy3 intensity; *P<0.05; **P<0.01; error bars represent ±1 s.d. Scale bar, 100 μm.
This particular experiment, however, also unveils the technical challenges to be overcome in our proposed approach. Concretely, the subsequent electrical measurements of hybridization by turning the role of each graphene into a sensing FET failed to show any clear-cut results as in the previous experiments (see Supplementary Fig. 6), which we may ascribe to the following. First, after the immobilization process, significant decrease of conductance was observed in the even-numbered graphene FETs; this is due likely to the Faradic currents that degrade the graphene FETs during the elongated immobilization time of 120 s, where this longer immobilization time is chosen to try to increase the amount of immobilized probe DNA. Second, the amount of Cy5-labelled probe DNA on the odd-numbered FETs was greatly reduced as a result of floating the even-numbered devices during immobilization, causing a decrease in the electric field magnitude around the graphene devices. Nonetheless, this experiment in Fig. 5 clearly demonstrates the possibility of immobilizing each graphene site with its own independent DNA sequence in an all-electrical manner.
Discussion
We have introduced 8-FET DNA sensor arrays fabricated from CVD graphene, which are aimed at a step towards realizing an electrical analogue of optical DNA microarray. Graphene engaged in the dual role of electrophoretic electrode and sensing FET has enabled site-specific operation of the graphene DNA array in an all-electrical manner. Its demonstrated target DNA detection sensitivity—100 fM in the passive immobilization case and 1 pM in the active site-specific immobilization case—is on a par with the ~1 pM sensitivity of the commercial optical DNA microarrays14,15,20—for example, AffyMetrix GeneChip15,20—that are being used for a broad palette of molecular diagnostic applications in both research and clinical settings. These applications include whole-transcript expression, drug metabolism and pharmacogenomics study, miRNA gene regulation, custom assays, cytogenetic analysis, genotyping and targeted resequencing. Therefore, if the number of graphene sensor sites in our array is substantially expanded in the future while maintaining the same sensitivity demonstrated here with a high yield, the graphene sensor array can occupy the same application space as the optical DNA microarray, while enjoying the label-free, chip-scale operation.
As mentioned in the introduction, certain high-end optical and electrochemical DNA sensors, such as those utilizing plasmonic, optical or chemical properties of gold nanoparticle labels21,22, those based on fibre-optic readout23,24 and those exploiting enzyme- or organometallic labels25,26,27,28, have achieved detection sensitivity orders of magnitude higher than our graphene sensor array. These ultra-sensitive sensors may open up new avenues of molecular diagnostic applications, which our graphene sensor array cannot enter with its present sensitivity. However, our sensor still fares well/better in sensitivity with most optical and electrochemical DNA sensors13, and is sensitive enough for the above-mentioned applications, while obviating the need for bulky optical readout and costly labelling steps21,22,23,24,25,26,27,28.
We have also demonstrated the challenges in site-specific immobilization of two different probe DNA sequences, while the site-specific immobilization of a single probe DNA sequence was successful. In particular, in the former effort, we have observed the loss of graphene conductance due to the more complex immobilization process and also the decrease in the amounts of immobilized probe DNA. These problems can in the future be addressed by optimization of the electrophoresis (for example, a.c. electrophoretic biasing, longer immobilization time but with a weaker bias) and structure (for example, independently addressable local reference electrodes).
Methods
Graphene device array fabrication
We transfer CVD graphene onto a 285-nm SiO2 layer thermally grown on top of a highly p-doped Si substrate, and pattern it into the 8 separate graphene sites by 60-watts O2 plasma at 200 mTorr for 60–70 s. We thermally evaporate 15/200/7-nm-thick Cr/Au/Cr layers to form the source and drain contacts to each graphene site, and passivate these metallic contacts by depositing a SiO2 layer via ALD (100 cycles at 250 °C for ~100 nm thickness). Graphene sites and wire-bonding regions are opened by 5:1 buffered oxide etchant. The graphene array device is then packaged with a polydimethylsiloxane microfluidic channel, which does not cover the wire-bonding openings but includes the graphene sites. The inlet and outlet of the microfluidic channel are connected with tubes for analyte injection and removal. The entire device consisting of the graphene array device and the microfluidic channel is attached onto a printed circuit board (PCB), and bonding wires provide electrical connections between the graphene array and the printed circuit board. An Ag/AgCl electrode inserted through one of the tubes to reach into the microfluidic channel serves as the reference electrode.
Analytes and DNA with their functionalization
Analytes used in our experiments are as follows: 1 mg ml−1 biotinylated BSA in nuclease-free water; 0.2 mg ml−1 streptavidin in 0.1 × PBS; probe DNA in 0.9 × PBS, target and control DNA in 1 × PBS for experiments in Fig. 2; probe DNA and target DNA in 0.005 × PBS for experiments in Fig. 3 (and Supplementary Fig. 4); two different sequences of probe DNA and their corresponding target DNA with each type in 0.005 × PBS for experiments in Fig. 5 (and Supplementary Figs 5 and 6). Here 1 × PBS is the standard PBS solution with 137 mM NaCl; 0.9 × PBS and 0.005 × PBS are diluted from 1 × PBS by 18.2 MΩ cm deionized water. The biotinylated BSA and streptavidin were purchased from Sigma Aldrich; DNA samples were purchased from Integrated DNA Technologies.
The probe DNA sequence forFigs 2 and 3 (and Supplementary Figs 3 and 4) is 5′-biotin-TGGCGACGGCAGCGAGGCTGAGGTAGTAGTTTGTACAGTTA-3′, with the italicized 20-base segment being complementary to the 20-mer target DNA inFigs 2 and 3 (and Supplementary Figs 3 and 4), whose sequence is 5′-CTGTACAAACTACTACCTCA-3′. This sequence is complementary to the 20-base segment of the 22-mer miRNA let7g, 5′-UGAGGUAGUAGUUUGUACAGUU-3′, where the segment is underlined. The expression level of miRNA let7g that is widely believed to mediate tumour suppression is closely associated with human cancer39,40, and the 20-base detection is sufficient in telling let7g apart from other miRNAs from the human genome47. The sequence of the control DNA in Fig. 2 is 5′-TGAGGTAGTAGATTGTATAGTT-3′. The two probe DNAs in Fig. 5 (and Supplementary Figs 5 and 6) are 5′-biotin-ACATGGTTGAAGTTTTCCTATTCCGGTGAGATGGAGGC AT-3′ and 5′-biotin-TCCATAACTGCTGGCTTAACGCCCATCTACACTTAGCTAT-3′, with their targets being 5′-ATGCCTCCATCTCACCGGAA-3′ and 5′-ATAGCTAAGTGTAGATGGGC-3′ (Supplementary Fig. 6). Labelling with Cy3 and Cy5 fluorophore for experiments in Figs 3 and 5 is done at either the 3′ or 5′ end.
The biotinylated BSA is introduced to the graphene array for all experiments for 2 h with the reference electrode, source and drain terminals grounded at room temperature. Streptavidin is introduced in Fig. 2 (and Supplementary Fig. 3) for 10 min with the reference electrode, source and drain terminals grounded. Streptavidin is introduced in Figs 3 and 5 (and Supplementary Figs 4–6) for 20 s with the noted bias conditions at room temperature. The probe DNA is allowed 10 min for immobilization in the experiments in Fig. 2, 20 s in Fig. 3 (and Supplementary Figs 4 and 5) and 120 s in Fig. 5 (and Supplementary Fig. 6) with the noted bias conditions at room temperature. Hybridization of target DNAs across all experiments is allowed for 4 h with the reference electrode, source and drain terminals grounded at room temperature.
Electrical measurement
IDS of each graphene site is amplified by a low-noise current preamplifier (SR570) and read by a digital multimeter (Agilent 34411 A) with a 10-mV sweep step of VREF. The value of V0 is extrapolated using a quadratic least-squares curve fit to the minimum 11 data points on the VREF-IDS curves, yielding a sub-10-mV accuracy of V0. The transient current across graphene sites is recorded using an oscilloscope (Textronix TDS 2024B) with the electrical pulse added by a waveform generator (Agilent 34401 A). A standard t-test is used to evaluate the statistical significance of the ΔV0 comparison between hybridization and control experiments (Fig. 2) and of the ΔV0 comparison between even- and odd-numbered graphene sites (Fig. 3; Supplementary Fig. 4).
Fluorescence measurement
Fluorescence images are taken using a laser scanning confocal microscope (Olympus FV300). A 534-nm laser with a 560–600-nm bandpass filter is used for Cy3 imaging, whereas a 633-nm laser with a 660-nm longpass and a 660–760-nm bandpass filters are used for Cy5 imaging. Fluorescence intensity of each graphene site is averaged on the graphene area exposed to DNAs. To compare the amount of immobilized DNAs across different experiments on the equal basis, Cy3 fluorescence data inFigs 3b and 5b (and Supplementary Figs 3 and 5) have been normalized to device 2 in Supplementary Fig. 3 by calibrating out the gain factor used in each imaging. Both fluorescent images and reflectance images of all eight graphene sites (taken with a 534-nm laser) are stitched together to form the combined array image with image contrast maximized to 5% saturation. A standard t-test is used to evaluate the statistical significance of the fluorescent intensity comparison between even- and odd-numbered graphene sites (Figs 3 and 5; Supplementary Fig. 5).
Additional information
How to cite this article: Xu, G. et al. Electrophoretic and field-effect graphene for all-electrical DNA array technology. Nat. Commun. 5:4866 doi: 10.1038/ncomms5866 (2014).
References
Cui, Y., Wei, Q., Park, H. & Lieber, C. M. Nanowire nanosensors for highly sensitive and selective detection of biological and chemical species. Science 293, 1289–1292 (2001).
Zheng, G., Patolsky, F., Cui, Y., Wang, W. U. & Lieber, C. M. Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nat. Biotech. 23, 1294–1301 (2005).
Chen, R. J. et al. Noncovalent functionalization of carbon nanotubes for highly specific electronic biosensors. Proc. Natl Acad. Sci. USA 100, 4984–4989 (2003).
Sorgenfrei, S. et al. Label-free single-molecule detection of DNA-hybridization kinetics with a carbon nanotube field-effect transistor. Nat. Nanotech. 6, 126–132 (2011).
Mannoor, M. S. et al. Graphene-based wireless bacteria detection on tooth enamel. Nat. Commun. 3, 763 (2012).
Park, J., Nam, S., Lee, M. & Lieber, C. M. Synthesis of monolithic graphene-graphite integrated electronics. Nat. Mater. 11, 120–125 (2012).
Jiang, S. et al. Real-time electrical detection of nitric oxide in biological systems with sub-nanomolar sensitivity. Nat. Commun. 4, 2225 (2013).
Liu, Y., Dong, X. & Chen, P. Biological and chemical sensors based on graphene materials. Chem. Soc. Rev. 41, 2283–2307 (2012).
Hahm, J. & Lieber, C. M. Direct ultrasensitive electrical detection of DNA and DNA sequence variations using nanowire nanosensors. Nano Lett. 4, 51–54 (2004).
Stern, E. et al. Label-free immunodetection with CMOS-compatible semiconducting nanowires. Nature 445, 519–522 (2007).
Novoselov, K. S. et al. A roadmap for graphene. Nature 490, 192–200 (2012).
Hess, L. H., Seifert, M. & Garrido, J. A. Graphene transistors for bioelectronics. Proc. IEEE 101, 1780–1792 (2013).
Sassolas, A., Leca-Bouvier, B. & Blum, L. J. DNA Biosensors and Microarrays. Chem. Rev. 108, 109–139 (2008).
MAQC Consortium. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161 (2006).
Dalma-Weiszhausz, D. D., Warrington, J., Tanimoto, E. Y. & Miyada, C. G. The Affymetrix GeneChip® platform: an overview. Methods Enzymol. 410, 3–28 (2006).
Colombo, L., Wallace, R. M. & Ruoff, R. S. Graphene growth and device integration. Proc. IEEE 101, 1536–1556 (2013).
Dong, X., Shi, Y., Huang, W., Chen, P. & Li, L. Electrical detection of DNA hybridization with single-base specificity using transistors based on CVD-Grown graphene sheets. Adv. Mater. 22, 1649–1653 (2010).
Chen, T. et al. Label-free detection of DNA hybridization using transistors based on CVD grown graphene. Biosens. Bioelectron. 41, 103–109 (2013).
Cai, B. et al. Ultrasensitive label-free detection of PNA-DNA hybridization by reduced graphene oxide field-effect transistor biosensor. ACS Nano 8, 2632–2638 (2014).
The AffyMetrix DNA Microarray website ( http://www.affymetrix.com/ Accessed 15 June 2014).
Yao, X. et al. Sub-attomole oligonucleotide and p53 cDNA determinations via a high-resolution surface plasmon resonance combined with oligonucleotide-capped gold nanoparticle signal amplification. Anal. Biochem. 354, 220–228 (2006).
Taton, T. A., Mirkin, C. A. & Letsinger, R. L. Scanometric DNA array detection with nanoparticle probes. Science 289, 1757–1760 (2000).
Song, L., Ahn, S. & Walt, D. R. Fiber-optic microsphere-based arrays for multiplexed biological warfare agent detection. Anal. Chem. 78, 1023–1033 (2006).
Bowden, M., Song, L. & Walt, D. R. Development of a microfluidic platform with an optical imaging microarray capable of attomolar target DNA detection. Anal. Chem. 77, 5583–5588 (2005).
Gao, Z. Q., Rafea, S. & Lim, L. H. Detection of nucleic acids using enzyme-catalyzed template-guided deposition of polyaniline. Adv. Mater. 19, 602–606 (2007).
Azek, F., Grossiord, C., Joannes, M., Limoges, B. & Brossier, P. Hybridization assay at a disposable electrochemical biosensor for the attomole detection of amplified human cytomegalovirus DNA. Anal. Biochem. 284, 107–113 (2000).
Takenaka, S., Yamashita, K., Takagi, M., Uto, Y. & Kondo, H. DNA sensing on a DNA probe-modified electrode using ferrocenylnaphthalene diimide as the electrochemically active ligand. Anal. Chem. 72, 1334–1341 (2000).
Xie, H., Zhang, C. & Gao, Z. Amperometric detection of nucleic acid at femtomolar levels with a nucleic acid/electrochemical activator bilayer on gold electrode. Anal. Chem. 76, 1611–1617 (2004).
Ghindilis, A. L. et al. CombiMatrix oligonucleotide arrays: genotyping and gene expression assays employing electrochemical detection. Biosens. Bioelectron. 22, 1853–1860 (2007).
Gurtner, C. et al. Microelectronic array devices and techniques for electric field enhanced DNA hybridization in low-conductance buffers. Electrophoresis 23, 1543–1550 (2002).
Wang, J., Cai, X., Wang, J., Jonsson, C. & Palecek, E. Trace measurements of RNA by potentiometric stripping analysis at carbon paste electrodes. Anal. Chem. 67, 4065–4070 (1995).
Wang, J., Zhang, X., Parrado, C. & Rivas, G. Controlled release of DNA from carbon-paste microelectrodes. Electrochem. Commun. 1, 197–202 (1999).
Li, X. et al. Transfer of large-area graphene films for high-performance transparent conductive electrodes. Nano Lett. 9, 4359–4363 (2009).
Reina, A. et al. Large area, few-layer graphene films on arbitrary substrates by chemical capor deposition. Nano Lett. 9, 30–35 (2009).
Cohen-Karni, T., Qing, Q., Li, Q., Fang, Y. & Lieber, C. M. Graphene and nanowire transistors for cellular interfaces and electrical recording. Nano Lett. 10, 1098–1102 (2010).
Hess, L. H. et al. Graphene transistor arrays for recording action potentials from electrogenic cells. Adv. Mater. 23, 5045–5049 (2011).
Ohno, Y., Maehashi, K., Yamashiro, Y. & Matsumoto, K. Electrolyte-gated graphene field-effect transistors for detecting pH and protein adsorption. Nano Lett. 9, 3318–3322 (2009).
Stern, E. et al. Importance of the Debye screening length on nanowire field effect transistor sensors. Nano Lett. 7, 3405–3409 (2007).
Ohshima, K. et al. Let-7 MicroRNA Family Is Selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 5, e13247 (2010).
Kumar, M. S. et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl Acad. Sci. USA 105, 3903–3908 (2008).
Star, A. et al. Label-free detection of DNA hybridization using carbon nanotube network field-effect transistors. Proc. Natl Acad. Sci. USA 103, 921–926 (2006).
Poghossian, A., Cherstvy, A., Ingebrandt, S., Offenhäusser, A. & Schöning, M. J. Possibilities and limitations of label-free detection of DNA hybridization with field-effect-based devices. Sens. Actuator B Chem. 111–112, 470–480 (2005).
Uslu, F. et al. Labelfree fully electronic nucleic acid detection system based on a field-effect transistor device. Biosens. Bioelectron. 19, 1723–1731 (2004).
Barbaro, M. et al. Fully electronic DNA hybridization detection by a standard CMOS biochip. Sens. Actuator B Chem. 118, 41–46 (2006).
Kataoka-Hamai, C. & Miyahara, Y. Label-free detection of DNA by field-effect devices. Sensors J. IEEE 11, 3153–3160 (2011).
Bard, A. J. & Faulkner, L. R. Electrochemical Methods—Fundamentals and Applications 2nd edn Wiley (2004).
National Institute of Health (NIH) BLAST tool for human genome search ( http://blast.ncbi.nlm.nih.gov/ Accessed 15 June 2014).
Acknowledgements
The authors are grateful to the support of this research by the Air Force Office of Scientific Research under contract FA9550-13-1-0211, the Office of Naval Research under contract N00014-13-1-0806, National Science Foundation under contract DMR-1231319 and Catalyst Foundation, Valhalla, NY. We thank Kok Hao Chen and Xiaowei Zhuang of Harvard University for help with DNA preparation.
Author information
Authors and Affiliations
Contributions
G.X., J.A. and D.H. conceived the project. K.Y.M.Y., Y.S. and J.K. prepared graphene samples. G.X. and J.A. designed the devices. G.X. fabricated the devices. G.X. and J.A. performed the electrical measurements and fluorescence microscopy. G.X., J.A., L.Q., H.Y. and D.H. analysed the data and wrote the paper. All authors discussed the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-6 and Supplementary Notes 1-4 (PDF 585 kb)
Rights and permissions
About this article

Cite this article
Xu, G., Abbott, J., Qin, L. et al. Electrophoretic and field-effect graphene for all-electrical DNA array technology. Nat Commun 5, 4866 (2014). https://doi.org/10.1038/ncomms5866
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5866
This article is cited by
-
Review on two-dimensional material-based field-effect transistor biosensors: accomplishments, mechanisms, and perspectives
Journal of Nanobiotechnology (2023)
-
Scalable graphene sensor array for real-time toxins monitoring in flowing water
Nature Communications (2023)
-
Micro- and nano-devices for electrochemical sensing
Microchimica Acta (2022)
-
Fabrication and microfluidic analysis of graphene-based molecular communication receiver for Internet of Nano Things (IoNT)
Scientific Reports (2021)
-
Printable graphene BioFETs for DNA quantification in Lab-on-PCB microsystems
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
