
Abstract
The nature and assembly of the chlamydial division septum is poorly defined due to the paucity of a detectable peptidoglycan (PG)-based cell wall, the inhibition of constriction by penicillin and the presence of coding sequences for cell wall precursor and remodelling enzymes in the reduced chlamydial (pan-)genome. Here we show that the chlamydial amidase (AmiA) is active and remodels PG in Escherichia coli. Moreover, forward genetics using an E. coli amidase mutant as entry point reveals that the chlamydial LysM-domain protein NlpD is active in an E. coli reporter strain for PG endopeptidase activity (ΔnlpI). Immunolocalization unveils NlpD as the first septal (cell-wall-binding) protein in Chlamydiae and we show that its septal sequestration depends on prior cell wall synthesis. Since AmiA assembles into peripheral clusters, trimming of a PG-like polymer or precursors occurs throughout the chlamydial envelope, while NlpD targets PG-like peptide crosslinks at the chlamydial septum during constriction.
Similar content being viewed by others


Introduction
The stress-bearing peptidoglycan (PG)-based cell wall protects bacterial cells from physical and chemical insults. PG (also known as murein) synthesis occurs throughout the envelope and at the division septum1,2. The septal PG along with constriction force by the cytokinetic ring assembled from FtsZ tubulin directs the envelope into an annular structure at the division plane to enable membrane fusion, ultimately compartmentalizing the dividing cell into two separate daughter chambers (Fig. 1a)3,4. The building block of PG is lipid II, an N-acetyl-glucosamine(GlcNAc)-N-acetyl-muramic acid (MurNAc)-pentapeptide unit carried by the phosphorylated isoprenoid bactoprenol (C55~\nP). During PG synthesis, the MurNAc-GlcNAc disaccharide units are polymerized into linear glycan strands by transglycosylase enzymes. The growing polymer is further fortified by crosslinking of the pentapeptide moieties by transpeptidases known as penicillin-binding proteins (PBPs; Fig. 1b). Upon synthesis of the septal PG and the ensuing compartmentalization, the septal PG is split to accommodate daughter cell separation, a task executed by PG remodelling enzymes such as lytic transglycosylases, amidases (N-acetylmuramoyl-L-alanine hydrolases) and peptidases (LD-carboxypeptidases and DD-endopeptidases) that act on the glycan, amide or peptide bonds in PG, respectively (Fig. 1a,b)3.

(a) Coordinated envelope constriction in E. coli and W. chondrophila, a member of the Chlamydiales order, by the cytokinetic machinery and (putative) cell wall biosynthetic and remodelling enzymes including amidases and the putative endopeptidase NlpD from W. chondrophila (NlpDWch). RodZWch was recently identified as an early recruit to the division plane, raising the possibility that it acts in an early cytokinetic event11. (b) Schematic of cell wall assembly pathway in E. coli and the predicted pathway of a PG-like cell wall in W. chondrophila, as well as the O-antigen pathway for biosynthesis of LPS in the E. coli outer membrane (OM). Note that both pathways use bactoprenol (C55~\nP) as membrane carrier for precursor assembly. The possible removal of the dissaccharide unit from PG or lipid II is indicated with a question mark and a transparent dissaccharide unit. (c) Gene organization of the region encompassing the coding sequences for AmiA and NlpD in W. chondrophila. Arrows indicate gene orientation in the W. chondrophila genome. (d) Predicted domain organization of W. chondrophila AmiA and NlpD. SS, signal sequence; LytC, (Pfam PF01520) amidase_3 domain; LysM, (Pfam PF01476) LysM-like PG-binding domain. Numbers refer to amino-acid positions in the translation product.
In addition to providing constriction force, FtsZ organizes septal PG synthesis and remodelling events1,2. Although most bacteria rely on FtsZ for division, several bacterial lineages, such as pathogens belonging to the phylum Chlamydiae, do not encode primary structural homologues of FtsZ in their genomes1. Thus, alternative organizers of PG synthesis/remodelling and of cytokinesis must exist. In principle, owing to their obligate intracellular life style and a protective network of proteins with disulphide bridges on the surface of elementary bodies (the infectious extracellular developmental stage), Chlamydiae should not need PG for protection from osmotic stress. Interestingly, however, despite the massive reduction in coding capacity of chlamydial genomes, a seemingly functional lipid II biosynthetic pathway along with several putative PG biosynthetic (transpeptidases, for example, FtsI) and predicted remodelling enzymes (putative amidases and endopeptidases) are encoded5,6,7,8. The latter suggests that chlamydial pathogens polymerize a septal PG derivative (or at least a PG remnant). Indeed, immunofluorescence labelling with antibodies to the Ribi adjuvant that contains mycobacterial cell wall skeleton or direct fluorescent labelling of a modified D-amino acid dipeptide revealed a non-proteinaceous PG-like substance or at least a dipeptide-derived PG precursor at the septum9,10. Moreover, penicillins (inhibitors of PBPs) block chlamydial division8,10,11. However, no coding sequences for known PG transglycosylation enzyme homologues are found in the genomes of chlamydial pathogens8, raising the intriguing possibility that this PG-like material lacks chains of glycan polymers and that instead the disaccharide units from lipid II remain unpolymerized after transpeptidation. Alternatively, unknown transglycosylation enzymes may promote glycan chain formation within the chlamydial cell wall. Modification of PG-like material, its synthesis in reduced amounts and/or its confinement in space or time could reflect an adaptation of chlamydial pathogens to the host by reducing the activation of NOD1/2 intracellular pattern recognition receptors that detect MurNAc-containing muropeptide fragments12.
Division in the absence of classical PG and FtsZ as that seen for the L-form bacteria and mycoplasmas belonging to the phylum Firmicutes occurs in an erratic and inefficient manner by membrane blebbing, budding or stretching1,13,14. By contrast, cell division in the phylum Chlamydiae is highly coordinated and regular, resembling the binary fission of cocci8,15. It is unknown how chlamydia execute division and if they remodel their septal PG-like material, but recently the first septal proteins of Chlamydiae have been identified11. Escherichia coli mutants lacking all three amidase paralogues offer a convenient system to probe for septal PG remodelling by amidase-like activities, as inactivation of the three amidase genes (amiA, amiB and amiC) prevents cell separation, yielding a chaining phenotype16. Interestingly, the barrier function of the outer membrane (conferred by lipopolysaccharide17 (LPS), which also uses C55~\nP as a carrier for the biosynthesis of its precursor; Fig. 1b) is compromised in the ΔamiA, ΔamiB; ΔamiC (henceforth ΔABC) triple mutant for reasons that are unclear18.
Here, using E. coli ΔABC as a surrogate host, we first confirmed that chlamydial AmiA orthologues restore cell separation and LPS barrier function, indicating that they are indeed active amidases. We then isolated a suppressive mutation in the E. coli gene encoding the NlpI lipoprotein that restores LPS function and alters the PG peptide crosslinking ratio in ΔABC cells. We provide evidence that chlamydial NlpD can bind PG in vitro and that it has PG peptidase activity in E. coli cells that are mutant for nlpI in vivo. Importantly, immunolocalization of dividing chlamydial (Waddlia chondrophila) cells unveils NlpD as the first septal cell-wall-binding protein and shows that it depends on a PG-like polymer for localization to the division septum. As AmiA is distributed in the cell envelope, our results support a model in which AmiA trims a PG-like polymer or lipid II throughout the envelope, while NlpD acts on peptide crosslinks at the division septum of human chlamydial pathogens.
Results
Activity and peripheral localization of chlamydial amidases
As chlamydial pathogens are typically small and difficult to grow, we exploited the robust growth and larger cell size of W. chondrophila, a member of the Chlamydiales order and a strict intracellular pathogen associated with bovine abortion and human miscarriage, for immunolocalization studies19,20. As it has so far not been possible to engineer targeted gene disruptions in W. chondrophila as for most members of the Chlamydiales, we complemented our cytological experiments with functional studies using E. coli as a surrogate host.
W. chondrophila AmiA (AmiAWch) is encoded in a gene cluster with predicted cell division and PG precursor (lipid II) biosynthesis enzymes (Fig. 1c). Akin to other chlamydial AmiA orthologues, AmiAWch exhibits 43% similarity (113/259) and 27% identity (70/259) to E. coli AmiA (AmiAEco), and features the predicted catalytic residues within the LytC/Amidase_3 signature domain of amidases (Fig. 1d, Supplementary Fig. 1 and Supplementary Table 1). Interestingly, primary structure predictions suggest that chlamydial amidases lack the autoinhibitory alpha-helix (Supplementary Fig. 1) that occludes substrate access of the E. coli amidases and that must first be displaced by a cognate amidase activator for the acquisition of full enzymatic activity21,22. This raises the possibility that the active site of the chlamydial amidase homologues could be in a constitutively open (active) state. AmiAWch was able to rescue the cell separation defect (chaining phenotype) of the E. coli ΔABC mutant (Fig. 2a and Supplementary Fig. 2A) akin to AmiAEco. However, a significant amount of cell debris (‘ghosts’; arrowheads in Fig. 2a and Supplementary Fig. 2A) accumulated in the cultures expressing AmiAWch, presumably reflecting lysed cells from ectopic (un-restrained) amidase activity of AmiAWch (Fig. 2a) that cannot be properly controlled by E. coli. In support of this, we observed by way of a LacZ-based lysis assay that AmiAWch liberated LacZ much more efficiently from cells compared with AmiAEco (Supplementary Fig. 2B). Cell ‘ghosts’ were not seen when AmiAWch derivatives with single, double and/or quadruple mutations in key catalytic residues (H55A, E70A, H124A and/or E194A; Supplementary Fig. 1) or AmiAEco were expressed (Fig. 2a), indicating that catalytic activity underlies the lysis phenotype. The catalytic mutants were also unable to support cell separation even though wild-type (WT) and most mutant AmiAWch derivatives accumulated to comparable steady-state levels as determined by immunoblotting using polyclonal antibodies to AmiAWch (Supplementary Fig. 2C,D). Finally, to determine whether these functional characteristics are also retained in AmiA orthologues from other members of the Chlamydiales, we conducted complementation experiments with a plasmid expressing the AmiA orthologue from Simkania negevensis (AmiASne) or from Parachlamydia acanthamoebae (AmiAPac)23 (Supplementary Fig. 1) and found that both are also active as amidases, inducing lysis and supporting cell separation and ghost cell formation in E. coli (Supplementary Figs 2B,E,F and 3A–C).

(a) DIC images of WT and ΔABC; ΔABC+Pvan-empty, -amiAWch, -[amiA-nlpD]Wch and -amiAEco. Arrowheads point to ghost cells; Scale bar, 4 μm. AmiA point mutants are highlighted in yellow. (b) Growth of WT and mutant E. coli strains on McConkey agar supplemented with 0.5% glucose (McCG). (c) ϕP1 titre in the indicated strains were calculated as described in Methods and is reported as the log10 fold change. Error bars show the s.d. Data are from three biological replicates. (d) Lysozyme sensitivity of the indicated E. coli strains. 0.25 ml of saturated cultures were added to 5 ml of LB top agar and plated, the indicated amount of lysozyme was then spotted on the cell overlay and incubated overnight at 30 °C. Differences in sensitivity are reported as the difference in size (diameter) of the inhibition halo (mm). Error bars show the s.d. Data are from three biological replicates. (e) Bacitracin sensitivity of the indicated E. coli strains. 250 microliters of saturated cultures were added to 5 ml of LB top agar and plated, the indicated amount of bacitracin was then spotted on the cell overlay and incubated overnight at 30 °C. Differences in sensitivity are reported as the difference in size (diameter) of the inhibition halo (mm). Error bars show the s.d.
Further evidence that the chlamydial amidases indeed have lytic activity came from expression of WT and mutant AmiAWch in the Gram-negative Alpha-proteobacterium Caulobacter crescentus that naturally grows in hypo-osmotic fresh water niches24 and is thus more prone to lysis when PG integrity is compromised. We observed that WT AmiAWch, but not mutant derivatives, induced rapid lysis upon shifting aerated (shaking) C. crescentus cultures to stasis (Fig. 3a). Moreover, high-performance liquid chromatography (HPLC) analysis of muropeptides liberated from purified cell wall sacculi that had been digested with the N-acetyl-muramidase mutanolysin revealed that AmiAWch induces the appearance of several muropeptide fragments in C. crescentus that are not present in the control samples (from cells harbouring the empty vector), with a commensurate reduction in other muropeptide species (Fig. 3b).

(a) Overnight cultures of C. crescentus harbouring different constructs were left static at 30 °C and OD600 nm were recorded every 90 min. Lysis occurred in strains carrying [Pvan−amiAWch] and [Pvan−(amiA-nlpD)Wch] while OD600 nm of strains carrying [Pvan−amiAWchH124A], [Pvan−amiAWchH55A E70A H124A E194A], [Pvan−nlpDWch] and pMT335 were not affected. Error bars show the s.d. Data are from three biological replicates. (b) Muropeptide analysis of Caulobacter crescentus cell walls in the indicated strains harbouring different constructs as indicated on the figure. Differences among HPLC profiles are highlighted by asterisks.
As E. coli amidase mutants have compromised LPS-dependent outer membrane barrier function, they are unable to grow on medium containing detergents, including the bile acid deoxycholate in McConkey agar18,25 (Fig. 2b). Moreover, LPS is the receptor for bacteriophage ϕP1 and the ΔABC mutant displays an increased resistance towards ϕP1 compared with WT cells (Fig. 2c). Surprisingly, expression of WT AmiAWch corrected these deficiencies as well, while the AmiAWch catalytic mutants were unable or substantially reduced in their ability to support these functions (Fig. 2b,c). The ΔABC mutant is also sensitive to exogenously applied lysozyme (a muramidase) or bacitracin (an antibiotic interfering with C55~\nP recycling through inhibition of the kinase BacA; Figs 1b and 2d,e). While expression of AmiAEco corrects all deficiencies of the ΔABC mutant (Fig. 2a–f), AmiAWch was unable to correct the bacitracin sensitivity of the ΔABC mutant. We attribute this to the ectopic (lytic) activity of AmiAWch that, in the absence of the autoinhibitory region that is found in amidases such as AmiAEco (Supplementary Fig. 1), leads to ‘ghost’ cell formation and an imbalance in PG precursors and/or bactoprenol (C55~\nP) derivatives. Such an imbalance in C55~\nP might sensitize cells to inhibitors of the bactoprenol recycling pathway such as bacitracin. Taken together, we conclude that despite the massive genome reduction during the evolution of Chlamydiae, the coding sequence of a functional and lytic amidase has been retained.
To confirm that AmiAWch is indeed expressed in dividing W. chondrophila cells, we measured the abundance of the amiAWch transcripts by reverse transcription quantitative (RT)–PCR post-infection (p.i.) of Vero host cells infected with W. chondrophila (Fig. 4a). We also raised polyclonal antibodies to AmiAWch and probed for the presence of AmiAWch by immunoblotting during growth of W. chondrophila (Supplementary Fig. 4). These experiments revealed the amiAWch transcript and the AmiAWch translation product to be detectable at all time points p.i. Moreover, we used the anti-AmiAWch antiserum for immunofluorescence microscopy of cells 24 h p.i. and observed AmiAWch in clusters in the cell envelope and occasionally at the septum in deeply constricted cells (Fig. 4b). While our functional and cytological analyses provide compelling evidence that the chlamydial amidases are functional, expressed and at the correct subcellular compartment to process a PG-like polymer (synthesized by the PBP transpeptidases, PBP2 and/or PBP3)5,6, we cannot rule out that AmiAWch acts directly on the PG building block lipid II, splicing off the MurNAc-GlcNAc disaccharide once lipid II is flipped onto the periplasmic face of the cytoplasmic membrane and polymerized by PBPs (Fig. 1b). In fact, the companion paper by Klöckner et al.26 demonstrates that Chlamydiae pneumoniae AmiA can cleave lipid II in vitro. We thus hypothesize that AmiA is constitutively active and can release the MurNAc-GlcNAc disaccharide unit from Lipid II and/or from a septal/peripheral PG-like polymer, even in the absence of a topological amidase activator3,4,22. It is also conceivable that chlamydial AmiA orthologs are important for bactoprenol (C55~\nP) recycling, which could be limiting due to ongoing LPS (O-antigen) precursor biosynthesis as in E. coli17(Fig. 1a).

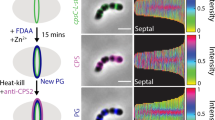
(a) Transcript levels of amiAWch and nlpDWch in Vero cells at different time points p.i. with W. chondrophila. Error bars show the s.d. Data are from three biological replicates. (b) NlpDWch localizes at the division plane (middle), while AmiAWch is localized at cell periphery with accumulation at constriction in dividing bacteria. Septal localization of NlpDWch is not affected by 4 h of penicillin or phosphomycin (phosph.) treatment (500 μg ml−1) administered 2 h p.i. Numbers indicate the fraction of cells with septal signal (n=100). Shown are merged images of cells stained with DAPI (blue), anti-Waddlia antibodies (red) and antibodies (green) to either AmiAWch (α-AmiAWch) or NlpDWch (α-NlpDWch). (c) Enrichment of NlpDWch at the division plane is not due to overlapping cells. Z-stacks were observed by confocal immunofluorescence microscopy. Reconstruction of a vertical cut through the cells is depicted here (inset). Quantification of pixel (px) intensities using ImageJ confirmed a concentration of NlpDWch at the division plane (right). (d) Immunofluorescence micrographs using antibodies to NlpDWch (red) and RodZWch (green) showing the delocalization of NlpDWch after 24 h of penicillin and phosphomycin administered 2 h p.i. Note that the midcell localization of RodZWch is maintained in the presence of penicillin, but not in the presence of phosphomycin as published previously11.
Amidases and endopeptidases influence LPS barrier function
Since E. coli amidases are required for proper LPS-dependent barrier function, they could modulate signalling by Toll-like innate immune receptors that detect bacterial cell envelope components such as LPS. To investigate how AmiAWch promotes LPS-dependent barrier function in E. coli ΔABC cells, we isolated a spontaneous ΔABC suppressor mutant (ΔABC nlpIW24STOP) that is able to grow on McConkey agar (supplemented with 0.5% Glucose; Fig. 5a). While this mutant exhibits near WT sensitivity to ϕP1 (Fig. 2c), the defects in cell separation, lysozyme and bacitracin sensitivity were not mitigated (Figs 2c,d and 5b). Thus, the LPS-dependent barrier function can be genetically uncoupled from cell separation.

(a) Effect of nlpI loss on growth on McConkey agar supplemented with 0.5% glucose (McCG) in WT and ΔABC strains. NlpI expression in ΔABC nlpIW24STOP prevents growth on McCG. Note that the presence of the vector appears to compromise growth of the nlpI mutant. (b) Representative DIC micrographs of ΔABCnlpIW24STOP harbouring the empty vector and a derivative carrying nlpI. Scale bar, 4 μm. (c) nlpI-dependent effect on McCG growth in different amidases mutant backgrounds, relative to WT and ΔABC. Deletion of nlpI in the ΔABC mutant restores the ability to grow on McCG on the indicated strains. (d) Deletion of nlpI in Δspr cells restores the ability to grow on McCG agar plates on the indicated strains.
Genome re-sequencing of this suppressor mutant disclosed a nonsense mutation of the tryptophan codon at position 24 (TGG→TAG) in the nlpI gene, encoding a tetratricopeptiderepeat (TPR)-containing lipoprotein required for virulence and adhesion in neonatal meningitis E. coli27,28. In support of the notion that the nlpIW24STOP mutation is a loss-of-function allele, expression of WT nlpI from a plasmid in the ΔABC nlpIW24STOP quadruple mutant again conferred the growth defect on McConkey agar medium typical of ΔABC cells (Fig. 5a). Conversely, deletion of nlpI (ΔnlpI::cmR) from ΔABC cells enabled growth of the resulting ΔABC ΔnlpI quadruple mutant on McConkey agar (Fig. 5c).
How might NlpI act? Since ΔABC nlpIW24STOP quadruple mutant cells are nearly as sensitive to bacitracin compared with the ΔABC parent, it is unlikely that NlpI acts in the bactoprenol (C55~\nP) recycling pathway (Fig. 1b). Interestingly, nlpI is known to interact genetically with spr, a gene encoding a DD-endopeptidase that cleaves the peptide cross-bridges between m-DAP and D-alanine in PG29. Moreover, loss-of-function mutations in nlpI or overproduction of the m-DAP-D-alanine DD-endopeptidase Pbp7 both suppress the conditional lethality of an spr deletion (Δspr) mutant30,31. Consistent with the notion that inactivation of nlpI affects and perhaps loosens septal PG, we observed that the poor growth of a Δspr strain on McConkey agar is attenuated by a ΔnlpI mutation (Fig. 5d), that ΔnlpI cells are sensitive towards increased expression of Spr or its paralogue YdhO (Fig. 6a) and that the inactivation of nlpI in WT or ΔABC cells resulted in an increased muro-tetrapeptide monomer to dimer ratio as determined by HPLC analysis (Table 1; Supplementary Fig. 5A,B). On the basis of these results, we propose that NlpI (perhaps via the TPR repeat) negatively regulates Spr and other endopeptidases that convert muro-tetrapeptide dimers to monomers. Thus, a proper balance of amidase and DD-endopeptidase activities governs the barrier function of LPS.

(a) Effect of overexpression of NlpDWch from pTrc99a (vector) phenocopies the effect of Spr and YdhO overproduction on plating efficiency of E. coli WT and ΔnlpI cells. Shown is a dilution series of the indicated strains. (b) Effect of overexpression of NlpDWch, NlpDSne or NlpDPac from Plac on pSRK on plating efficiency of WT and ΔnlpI E. coli cells. Shown is a dilution series of cells carrying pSRK (vector) derivatives expressing NlpDWch from Plac plated on LB with or without inducer (1 mM IPTG) of the indicated strains. (c) Lytic activity of NlpDWch, NlpDSne or NlpDPac expressed from Plac on pSRK in WT and ΔnlpI E. coli cells carrying the LacZ-expressing plasmid pLac290-Pconst::lacZ (used because the E. coli parent, TB28, is lacZ minus). Beta-galactosidase activities were measured on SN of induced and non-induced cultures of WT E. coli carrying Plac-nlpD constructs. Error bars show the s.d. Data are from three biological replicates. (d) Binding of different His6-NlpDWch variants to E. coli murein (PG) sacculi. Purified WT and mutant NlpDWch (3 μg each) were incubated with or without 1 mg of E. coli sacculi. Sacculi were pelleted by ultracentrifugation and washed once with buffer. Immunoblotting with antibodies to NlpDWch was used to reveal NlpDWch in the supernatant (S), the wash fraction (W) or the pellet (P) fraction. The size markers (in kDa) are indicated on the right. The arrow on the left denotes the position of His6-NlpDWch.
Septal localization and cell wall remodelling by NlpDWch
Prompted by these functional interactions between the amidases and DD-endopeptidases in E. coli, we searched for putative DD-endopeptidases encoded in the W. chondrophila genome using E. coli Spr, YdhO and YebA as BLASTP queries. After considering the genomic context where the candidates are encoded, we focused our attention on the gene annotated as nlpD (nlpDWch). nlpDWch is embedded within a cluster of genes predicted to code for cell wall and division functions (Fig. 1c). Although it is difficult to predict from the primary structure if the NlpDWch translation product has endopeptidase activity, the presence of two putative LysM domains (Fig. 1d; Supplementary Fig. 6) that are known to mediate PG-binding and/or septal localization in other proteins32 along with a 36% similarity with NlpDEco (Supplementary Fig. 6) make it a strong candidate to act in remodelling of a PG-like polymer and/or chlamydial division.
Having shown that the E. coli ΔnlpI::cmR cells are a suitable background in which to probe for endopeptidase activity, we then tested whether expression of NlpDWch reduces the plating efficiency of ΔnlpI::cmR cells, akin to expression of Spr or YdhO, without affecting WT cells (Fig. 6a). Indeed, WT NlpDWch but not mutant variants harbouring missense mutations in conserved residues within the LysM domain (Supplementary Fig. 6) caused a strong reduction in plating efficiency of ΔnlpI::cmR cells compared with WT cells (Supplementary Fig. 7A). Several of these missense mutants accumulated NlpDWch to similar levels as WT (Supplementary Fig. 7B), suggesting that conserved residues in the LysM domain are required for function. A similar reduction in plating efficiency was observed upon expression of the NlpD orthologue from Simkania negevensis or Parachlamydia acanthamoebae (NlpDSne or NlpDPac, respectively) in ΔnlpI::cmR cells, but not in WT E. coli (Fig. 6b). Moreover, induction of NlpDWch, NlpDSne or NlpDPac expression caused an efficient release of LacZ from E. coli ΔnlpI::cmR cells and only poorly from WT E. coli cells (Fig. 6c). To confirm that NlpDWch can affect E. coli PG, we conducted HPLC analysis of muropeptides released from sacculi of ΔABC cells expressing NlpDWch. This revealed a similar increase in muro-tetrapeptide monomer to dimer ratio (Table 1; Supplementary Fig. 5A,B) compared with the empty vector, as that resulting from the loss of NlpI. Interestingly, the increase in tetrapeptide monomer to dimer ratio was mitigated upon co-expression of AmiAWch with NlpDWch (Table 1; Supplementary Fig. 5), despite near-identical steady-state levels of NlpDWch in cells with the AmiAWch-NlpDWch co-expression compared with cells with the NlpDWch single expression plasmid (Supplementary Fig. 2C), suggesting that AmiAWch is epistatic over NlpDWch and, thus, that they act in the same pathway.
Next, we explored the expression and localization of NlpDWch in W. chondrophila grown in Vero cells. RT–PCR (Fig. 4a) and immunoblotting using polyclonal antibodies to NlpDWch (Supplementary Fig. 4) showed that NlpDWch is indeed expressed. Importantly, IFM performed on Vero cells 24 h p.i. with W. chondrophila revealed fluorescent bands of NlpDWch at midcell in 50.8±1.1% of constricted cells (Fig. 4b) and it can already be seen at the septum early during constriction (in 47% of the cells, Supplementary Fig. 8), with a significant increase in frequency at the later stages of constriction. Quantitative analysis of the fluorescence traces from NlpDWch, DAPI (4′,6-diamidino-2-phenylindole)-stained chromosome and the anti-Waddlia-stained cell envelope revealed a sharp increase in NlpDWch abundance at the medial site, in between two broad peaks of DAPI-stained DNA flanked by the cell envelope (Fig. 4c). This septal localization is still maintained for 4 hours after inhibition of division with penicillin, an inhibitor of PG transpeptidation enzymes (Pbp2/3), or with phosphomycin, an inhibitor of the lipid II biosynthetic enzyme MurA (Fig. 4b). However, 20 h later only peripheral NlpDWch was observed under both conditions (septal only in 4.4% and 2.4% of dividing cells, respectively; Fig. 4d and Supplementary Fig. 8). By contrast, the early cell division marker RodZWch was still septal in the presence of the transpeptidation inhibitor (septal in 43.4% of dividing cells), but not when lipid II biosynthesis is blocked (Fig. 4d), as reported recently11.
As these findings suggest that chlamydial NlpD is recruited to the division septum by its substrate, a PG-like D-amino acid-containing peptide polymer, we used a pelleting assay with intact and purified E. coli polymeric PG (sacculi) to determine whether purified WT or mutant His6-tagged NlpDWch (His6-NlpDWch) can indeed bind polymeric PG in vitro (Fig. 6d). E. coli sacculi pulled down WT His6-NlpDWch, but not mutant derivative lacking two conserved residues in the LysM domain (N213A/D214A; Supplementary Fig. 6), indicating that NlpDWch can bind PG directly.
Discussion
Our data support a model in which NlpDWch recognizes a PG-like polymer at the division septum, while AmiA trims this polymer or lipid II molecules throughout the envelope and possibly at the division furrow in the final stages of division, perhaps to lower NOD1/2-inducing MurNAc-peptide signals12,33 during chlamydial infections, akin to the staphylococcal autolysins that prevent detection by the Drosophila innate immune system33. Interestingly, a PG-like polymer was recently extracted from the ‘environmental’ chlamydia Protochlamydia amoebophila, an amoebal symbiont, but similar attempts were unsuccessful for Simkania negevensis34. Nevertheless, we found that the S. negevensis genome encodes functional AmiA and NlpD (AmiASne and NlpDSne) that are active on E. coli PG, suggesting that PG-like material is also present in S. negevensis. It is possible that pervasive PG synthesis throughout the envelope (giving rise to intact sacculi) is a feature of chlamydial lineages that establish symbiotic relationships with amoebae, while chlamydial human pathogens only produce a cryptic, modified, short-lived, thin and/or spatially restricted PG. In light of the recent evidence that PG synthesis can indeed occur de novo (that is, in the absence of a preexisting template) in Bacillus subtilis cells35, it is conceivable that a PG-like structure is confined temporally and spatially to the division septum in chlamydial pathogens. Recent experiments using fluorescently labelled D-amino-dipeptides provided evidence of a septal peptide component in PG (or in lipid II) of Chlamydia trachomatis10, in support of the earlier discovery of the SEP antigen (recognized by antibodies raised against the mycobacterial cell wall containing RIBI adjuvant) at the division septum of C. trachomatis and Chlamydia psittaci9. Our data indicate that a septal PG-like polymer in the human chlamydial pathogen W. chondrophila is both a substrate and an important localization cue for a septal PG-binding protein (NlpD) during chlamydial cell division. Moreover, using purified components Klöckner et al.26 provide compelling biochemical evidence that C. pneumoniae NlpD has carboxypeptidase activity in vitro.
The notion that human chlamydial pathogens rely on a PG-like peptide polymer at the division site and that they localize NlpD to this site in the absence of an FtsZ homologue raises the important evolutionary question how a PG-like division septum is positioned in different bacteria. Our recent identification of the RodZ homologue RodZWch as an early septal protein11 along with the finding reported here that RodZWch is still septal under conditions when NlpDWch is dispersed (in the presence of penicillin) suggest that RodZWch localizes to the septum before NlpDWch and that it could play a key role in orchestrating septal assembly (via PBPs) and dissolution (via NlpDWch and possibly AmiAWch). While FtsZ is known to be dispensable in another bacterial phylum (the Firmicutes) when PG is absent13,14, the only known case of FtsZ-independent PG-based septation has been described for filamentous bacteria from the phylum Actinobacteria (genus Streptomyces) that rely on FtsZ exclusively for septation during spore development, while crosswalls formed during vegetative (hyphal) growth do not require FtsZ36. As the Streptomyces do not encode an obvious RodZ orthologue in their genomes, different solutions have emerged for the synthesis of a PG-based septum and the subsequent recruitment of remodelling enzymes in diverse bacterial phyla and even in eukaryotic organelles1,37.
Methods
Bacterial strains and growth conditions
Strains and plasmids used in this study are listed in Supplementary Table 2 and their constructions are described in the Supplementary Methods section. E. coli strains were grown at 30 °C in Luria–Bertani (LB) broth, LB-agar38 or McConkey agar supplemented with gentamycin (10 μg ml−1), IPTG (isopropyl-β-thio-galactopiranoside, 1 mM), as needed or otherwise indicated. C. crescentus strains were grown at 30 °C in peptone yeast extract supplemented with gentamycin (1 μg ml−1) as needed. Plasmids were introduced in E. coli by electroporation, chemical transformation or conjugation. W. chondrophila ATCC VR-1470T was grown in Vero cells as previously described5. Overnight cell cultures containing originally 105 cells ml−1 were infected with a 2,000 × dilution of W. chondrophila. The cells were then centrifuged for 15 min (to improve contact of W. chondrophila) at 1,790g, incubated 15 min at 37 °C and washed with PBS before addition of fresh media.
Differential interference contrast microscopy
Cultures were grown at 30 °C in LB medium unless otherwise indicated. Unless otherwise indicated cells were imaged by DIC (Differential Interference Contrast) optics on microscope slides harbouring a thin (1%) agarose pad. A Zeiss Axioplan 2 microscope fitted with an HQ Snapshot camera, a Zeiss oil immersion objective ( × 100/1.45 numerical aperture) were used to acquire DIC images using software from Metamorph (Universal Imaging). Cells and ghosts were quantified using ImageJ software ( http://rsbweb.nih.gov/ij/).
Immunofluorescence and confocal microscopy
Infected Vero cells on coverslips were fixed with ice-cold methanol for 5 min at room temperature. Infection rate, inclusions and aberrant bodies were quantified by fluorescence microscopy by counting a minimum of hundred cells in duplicate5. Images were taken by confocal microscopy using a Zeiss LSM 510 Meta (Zeiss, Oberkochen, Germany). Images were then treated and quantified using ImageJ software.
AmiA and NlpD purification and production of antibodies
His6-NlpDWch protein and the N213A/D214A mutant derivative were expressed from pET28a in E. coli Rosetta (DE3)/pLysS (Novagen, Madison, WI) and purified under native conditions using Ni2+ chelate chromatography. A 5 ml overnight culture was diluted into 1 l of pre-warmed LB. OD600 nm were monitored until OD600 nm=~\n0.3–0.4, then 1 mM IPTG was added to the culture and growth continued. After 3 h cells were pelleted, and resuspended in 25 ml of lysis buffer (10 mM Tris HCl (pH 8), 0.1 M NaCl, 1.0 mM β-mercaptoethanol, 5% glycerol, 0.5 mM imidazole Triton X-100 0.02%). Cells were sonicated (Sonifier Cell Disruptor B-30; Branson Sonic Power. Co., Danbury, CT) on ice using 12 bursts of 20 s at output level 5.5. After centrifugation at 4,300 g for 20 min, the supernatant was loaded onto a column containing 5 ml of Ni-NTA agarose resin pre-equilibrated with lysis buffer. Column was rinsed with lysis buffer, 400 mM NaCl and 10 mM imidazole, both prepared in lysis buffer. Fractions were collected (in 300 mM Imidazole buffer, prepared in lysis buffer) and used to immunize New Zealand white rabbits (Josman LLC, Napa, CA).
His6-SUMO-AmiAWch was expressed from pCWR547-amiAWchover in E. coli Rosetta (DE3)/pLysS and purified in denaturing buffer (8 M Urea, 100 mM NaH2PO4, 25 mM Tris). A 5 ml overnight culture was diluted into 1L of pre-warmed LB. OD600 nm was monitored until OD600 nm=~\n0.3–0.4, then 1 mM IPTG was added to the culture transferred at room temperature for 5 h. Thereafter, cells were pelleted, resuspended in 25 ml of lysis buffer (10 mM Tris HCl (pH 8), 0.1 M NaCl, 1.0 mM β-mercaptoethanol, 5% glycerol, 0.5 mM imidazole, Triton X-100 0.02%). Cells were sonicated (Sonifier Cell Disruptor B-30; Branson Sonic Power. Co., Danbury, CT) on ice using 12 bursts of 20 s at output level 5.5. After centrifugation at 4,300 g for 20 min, the supernatant were discarded and the pellet resuspended in 25 ml of Buffer B (denaturing buffer, pH 8.0), then centrifuged at 4,300 g for 20 min, the supernatant was loaded onto a column containing 5 ml of Ni-NTA agarose resin. Column was rinsed with Buffer B, Buffer C (denaturing buffer, pH 6.3) and eluted with Buffer E (denaturing buffer, pH 4.5). Fractions were collected, the protein was excised from a 15% SDS polyacrylamide gel and used to immunize New Zealand white rabbits (Josman LLC, Napa, CA).
Immunoblots
Pelleted cells were resuspended in 1 × SDS sample buffer (50 mM Tris–HCl (pH 6.8), 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.02% Bromophenol Blue), heated to 95 °C for 10 min and stored at −20 °C. The resulting cell extracts were resolved on SDS–PAGE gels and transferred onto PVDF (polyvinylidenfluoride) membranes.
PVDF membranes (Merck Millipore Headquarters, Billerica, MA) were blocked with TBS, 0.05% Tween 20 and 5% dry milk for 1 h and then incubated for 1 h with the primary antibodies diluted in TBS, 0.05% Tween 20 and 5% dry milk. The different antisera (custom produced, as described above) were used at the following dilutions: anti-AmiAWch (1:10,000), anti-NlpDWch (1:10,000). The membranes were washed four times for 5 min in TBS and incubated 1 h with the secondary antibody (HRP-anti-Rabbit 1:10,000) diluted in TBS, 0.05% Tween 20 and 5% dry milk. The membranes were finally washed again four times for 5 min in TBS and revealed with Immobilon Western Blotting Chemoluminescence HRP substrate (Merck Millipore Headquarters, Billerica, MA).
Phage manipulation, lysozyme and antibiotic sensitivity tests
The bacterial strains used in the present study were used to produce ΦP1 lysates and tested for phage P1 sensitivity38. Saturated cultures of E. coli TB28 (ΔlacZYA<>frt) were used to produce ΦP1 lysates. Cells from overnight cultures were diluted 1:100 in LB with 25 mM CaCl2 and infected with ΦP1 lysate. Phage titres were calculated by spot and plating methods. Briefly 1.5 ml of cells from overnight cultures were pelleted and resuspended in 0.3 ml of LB with 5 mM of CaCl2, and then incubated for 10 min at 37 °C. A total of 0.05 ml of ΦP1 were added to the suspension and incubated at 37 °C for 20 min. Serial dilution of the suspension were made and added to 4 ml of LB top agar supplemented with 5 mM CaCl2, plates were then incubated overnight at 37 °C or until plaques are visible. The titre of the phage was calculated as plaque-forming units (pfu) per ml, then percentage of the titre relative to WT E. coli was calculated and reported as the log10 of the percentage. Values reported in the figures come from three independent biological replicas.
Sensitivity of bacterial strains presented in this study were determined as follows: 5 mg of lysozyme (AppliChem) and bacitracin (Sigma), respectively, were spotted on LB top agar containing 0.250 ml of saturated overnight cultures of the strains to be tested and incubated overnight at 30 °C. Diameter of the inhibition halo was measured from three biological replicates.
Muropeptide analysis
Chemical composition of PG was determined by the standardized procedure developed by Cecolabs (Tübingen, Germany). For PG isolation and characterization the strains were grown overnight in LB or peptone yeast extract media unless otherwise indicated. Cells were harvested by centrifugation (4,300 g, 20 min at 4 °C), supernatant was discarded and cells resuspended in 20 ml of phosphate buffered saline (pH 7.4; 1x PBS) and then boiled for 15 min, the resulting extract was centrifuged and after discarding the supernatant the pellet was stored at 4 °C.
Briefly, cell pellets were resuspended and boiled in SDS, cell wall material was harvested by centrifugation and broken with glass beads. Broken cell wall was digested with mutanolysin and analysed by HPLC39. HPLC analyses were performed with an Agilent 1200 system with a Prontosil C18-RP column (Bischoff Chromatography, Leonberg, Germany)40. Selected peaks were identified by matrix-assisted laser desorption ionization and electrospray mass spectrometry.
Quantitative analysis of selected peaks was done by integration of the peak area using the trapezoidal rule. The area of each peak was then used to derive the ratios of cell wall components among the different strains.
Murein (sacculi) pull-down assay
NlpDWch-His6 and NlpDWchN213A D214A-His6 were overproduced in E. coli Rosetta (λDE3)/pLys. Proteins were purified by nickel affinity chromatography as described above, concentrated by ultrafiltration in Amicon 3K columns (Millipore) and stored at −80 °C in Binding buffer (20 mM Tris–HCl, 1 mM MgCl2 30 mM NaCl, 0.05% Triton X-100, pH 6.8) containing 50% glycerol. Bradford assay was used to determine the protein concentration in each sample. E. coli murein (sacculi) was purchased from Cecolabs (Tuebingen, Germany) and resuspended in binding buffer at a concentration of 10 mg ml−1. Proteins (3 μg or 6 μg) were added to 1 mg murein in a total volume of 100 μl and incubated on ice for 30 min. Murein from samples was collected by centrifugation in a Beckman SW55Ti rotor at 303,648 g for 30 min at 4 °C. Sedimented murein was resuspended in 0.1 ml of cold binding buffer and centrifuged again. Murein pellets were resupended in 0.02 ml of cold binding buffer. The supernatant of the first centrifugation step (S), the supernatant of the washing step (W) and the pellet (P) were analysed by SDS–PAGE followed by immunoblot with anti-NlpD antiserum (see immunoblots for details).
Assay to determine the lytic activity of chlamydial amidases
WT and ΔABC E. coli cells harbouring pSRK (Plac−) and pMT335 (Pvan−) amidase plasmids were transformed with the low copy plasmid pLac290 harbouring a promoter of C. crescentus, which is constitutively active in E. coli (pLac290-Pconst::lacZ). Overnight cultures were diluted in fresh LB and incubated at 30 °C for 6 h. Culture supernatants (SN) were collected by centrifugation at 20,000 g for 5 min at room temperature. SN were diluted 1:10 in 1 ml of LB and 0.2 ml of diluted SN were used for standard β-Galactosidase (LacZ) assay38.
Quantitative real-time PCR
Infection of Vero cells by W. chondrophila was quantified by quantitative PCR11. At different time points after infection, infected cells were resuspended by scratching. Genomic DNA was extracted from 50 μl of cell suspension using the Wizard SV Genomic DNA purification system (Promega, Madison, WI). Elution was processed with 200 μl of water. Quantitative PCR was performed using iTaq supermix with ROX (BioRad, Hercules, CA). To detect W. chondrophila, 200 nM of primers WadF4 and WadR4, 100 nM of probe WadS2 and 5 μl of DNA were used. Cycling conditions were 3 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C for both PCRs. A StepOne Plus Real-time PCR System (Applied Biosystems, Carlsbad, CA) was used for amplification and detection of the PCR products.
Additional information
How to cite this article: Frandi, A. et al. FtsZ-independent septal recruitment and function of cell wall remodelling enzymes in chlamydial pathogens. Nat. Commun. 5:4200 doi: 10.1038/ncomms5200 (2014).
References
Margolin, W. FtsZ and the division of prokaryotic cells and organelles. Nat. Rev. Mol. Cell Biol. 6, 862–871 (2005).
Adams, D. W. & Errington, J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 7, 642–653 (2009).
Uehara, T. & Bernhardt, T. G. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr. Opin. Microbiol. 14, 698–703 (2011).
Erickson, H. P., Anderson, D. E. & Osawa, M. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol. Mol. Biol. Rev. 74, 504–528 (2010).
Bertelli, C. et al. The Waddlia genome: a window into chlamydial biology. PLoS ONE 5, e10890 (2010).
Stephens, R. S. et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759 (1998).
Henrichfreise, B. et al. Functional conservation of the lipid II biosynthesis pathway in the cell wall-less bacteria Chlamydia and Wolbachia: why is lipid II needed? Mol. Microbiol. 73, 913–923 (2009).
Ghuysen, J.-m., Goffin, C. & Inge, C. Lack of cell wall peptidoglycan versus penicillin sensitivity: new insights into the chlamydial anomaly. Antimicrob. Agents Chemother. 43, 2339–2344 (1999).
Brown, W. J. & Rockey, D. D. Identification of an antigen localized to an apparent septum within dividing Chlamydiae. Infect. Immun. 68, 708–715 (2000).
Liechti, G. W. et al. A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature 506, 507–510 (2014).
Jacquier, N., Frandi, A., Pillonel, T., Viollier, P. & Greub, G. Cell wall precursors are required to organize the chlamydial division septum. Nat. Commun. 5, 3578 (2014).
Underhill, D. M. Collaboration between the innate immune receptors dectin-1, TLRs, and Nods. Immun. Rev. 219, 75–87 (2007).
Leaver, M., Dominguez-Cuevas, P., Coxhead, J. M., Daniel, R. A. & Errington, J. Life without a wall or division machine in Bacillus subtilis. Nature 457, 849–853 (2009).
Lluch-Senar, M., Querol, E. & Pinol, J. Cell division in a minimal bacterium in the absence of ftsZ. Mol. Microbiol. 78, 278–289 (2010).
Pinho, M. G., Kjos, M. & Veening, J. W. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat. Rev. Microbiol. 11, 601–614 (2013).
Uehara, T., Parzych, K. R., Dinh, T. & Bernhardt, T. G. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 29, 1412–1422 (2010).
Ruiz, N., Kahne, D. & Silhavy, T. J. Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat. Rev. Microbiol. 7, 677–683 (2009).
Heidrich, C., Ursinus, A., Berger, J., Schwarz, H. & Höltie, J. V. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J. Bacteriol. 184, 6093–6099 (2002).
Baud, D. et al. Waddlia chondrophila: from bovine abortion to human miscarriage. Clin. Infect. Dis. 52, 1469–1471 (2011).
Baud, D. et al. Role of Waddlia chondrophila placental infection in miscarriage. Emerg. Infect. Dis. 20, 460–464 (2014).
Yang, D. C. et al. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc. Natl Acad. Sci. USA 108, E1052–E1060 (2011).
Yang, D. C., Tan, K., Joachimiak, A. & Bernhardt, T. G. A conformational switch controls cell wall-remodelling enzymes required for bacterial cell division. Mol. Microbiol. 85, 768–781 (2012).
Greub, G. et al. High throughput sequencing and proteomics to identify immunogenic proteins of a new pathogen: the dirty genome approach. PLoS ONE 4, e8423 (2009).
Poindexter, J. S. The caulobacters: ubiquitous unusual bacteria. Microbiol. Rev. 45, 123–179 (1981).
Tamaki, S. & Matsuhashi, M. Increase in sensitivity to antibiotics and lysozyme on deletion of lipopolysaccharides in Escherichia coli strains. J. Bacteriol. 114, 453–454 (1973).
Klöckner, A. et al. AmiAis a penicillin target enzyme with dual activity in the intracellular pathogen Chlamydia pneumoniae. Nat. Commun. 5, 4201 (2014).
Teng, C. H. et al. NlpI contributes to Escherichia coli K1 strain RS218 interaction with human brain microvascular endothelial cells. Infect. Immun. 78, 3090–3096 (2010).
Barnich, N., Bringer, M. A., Claret, L. & Darfeuille-Michaud, A. Involvement of lipoprotein NlpI in the virulence of adherent invasive Escherichia coli strain LF82 isolated from a patient with Crohn's disease. Infect. Immun. 72, 2484–2493 (2004).
Singh, S. K., SaiSree, L., Amrutha, R. N. & Reddy, M. Three redundant murein endopeptidases catalyse an essential cleavage step in peptidoglycan synthesis of Escherichia coli K12. Mol. Microbiol. 86, 1036–1051 (2012).
Hara, H., Abe, N., Nakakouji, M., Nishimura, Y. & Horiuchi, K. Overproduction of penicillin-binding protein 7 suppresses thermosensitive growth defect at low osmolarity due to an spr mutation of Escherichia coli. Microb. Drug Resist. 2, 63–72 (1996).
Tadokoro, A. et al. Interaction of the Escherichia coli lipoprotein NlpI with periplasmic Prc (Tsp) protease. J. Biochem. 135, 185–191 (2004).
Buist, G., Steen, A., Kok, J. & Kuipers, O. P. LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol. Microbiol. 68, 838–847 (2008).
Atilano, M. L. et al. Bacterial autolysins trim cell surface peptidoglycan to prevent detection by the Drosophila innate immune system. eLife 3, e02277 (2014).
Pilhofer, M. et al. Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ. Nat. Commun. 4, 2856 (2013).
Kawai, Y., Mercier, R. & Errington, J. Bacterial cell morphogenesis does not require a preexisting template structure. Curr. Biol. 24, 863–867 (2014).
McCormick, J. R. Cell division is dispensable but not irrelevant in Streptomyces. Curr. Opin. Microbiol. 12, 689–698 (2009).
Miyagishima, S. Y., Kabeya, Y., Sugita, C., Sugita, M. & Fujiwara, T. DipM is required for peptidoglycan hydrolysis during chloroplast division. BMC Plant Biol. 14, 57 (2014).
Miller, J. H. Experiment in Molecular Genetics Cold Spring Harbor Laboratory (1972).
de Jonge, B. L., Chang, Y. S., Gage, D. & Tomasz, A. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 267, 11248–11254 (1992).
Ute Bertsche, S.-J. Y. et al. Increased cell wall teichoic acid production and D-alanylation are common phenotypes among daptomycin-resistant methicillin-resistant Staphylococcus aureus (MRSA) clinical isolates. PLoS ONE 8, e67398 (2013).
Acknowledgements
Funding support was from the Fondation Leenaards, the Swiss National Science Foundation (CRSII3_141837) and the Canton de Genève. We thank Tsuyoshi Uehara, Tom Bernhardt, Manjula Reddy and Patrice Moreau for materials; Tsuyoshi Uehara, Tom Bernhardt, Martin Thanbichler and Miguel Valvano for helpful suggestions and Beate Henrichfreise for communicating unpublished results.
Author information
Authors and Affiliations
Contributions
A.F., N.J., G.G. and P.H.V. conceived and designed the experiments. A.F., N.J. and L.T. performed the experiments. A.F., N.J., G.G. and P.H.V. analysed the data. A.F., N.J., G.G. and P.H.V. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8, Supplementary Tables 1-2, Supplementary Methods and Supplementary References (PDF 1644 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Frandi, A., Jacquier, N., Théraulaz, L. et al. FtsZ-independent septal recruitment and function of cell wall remodelling enzymes in chlamydial pathogens. Nat Commun 5, 4200 (2014). https://doi.org/10.1038/ncomms5200
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5200
This article is cited by
-
Amidase activity is essential for medial localization of AmiC in Caulobacter crescentus
Current Genetics (2018)
-
Chlamydia cell biology and pathogenesis
Nature Reviews Microbiology (2016)
-
AmiA is a penicillin target enzyme with dual activity in the intracellular pathogen Chlamydia pneumoniae
Nature Communications (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
