
Abstract
Three-electron σ-bonding that was proposed by Linus Pauling in 1931 has been recognized as important in intermediates encountered in many areas. A number of three-electron bonding systems have been spectroscopically investigated in the gas phase, solution and solid matrix. However, X-ray diffraction studies have only been possible on simple noble gas dimer Xe∴Xe and cyclic framework-constrained N∴N radical cations. Here, we show that a diselena species modified with a naphthalene scaffold can undergo one-electron oxidation using a large and weakly coordinating anion, to afford a room-temperature-stable radical cation containing a Se∴Se three-electron σ-bond. When a small anion is used, a reversible dimerization with phase and marked colour changes is observed: radical cation in solution (blue) but diamagnetic dimer in the solid state (brown). These findings suggest that more examples of three-electron σ-bonds may be stabilized and isolated by using naphthalene scaffolds together with large and weakly coordinating anions.
Similar content being viewed by others

Introduction
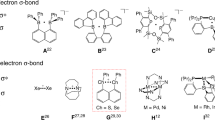
Odd-electron species exist as intermediates in many chemical reactions and play an important role in bond formation and cleavage processes1,2,3,4,5,6,7. Odd-electron bonding is of both fundamental and practical interest8,9,10,11,12,13. Three-electron σ-bonding was first proposed by Linus Pauling in 1931 (ref. 14) and has been recognized as important in major intermediates encountered in many areas such as radical chemistry, biochemistry, organic reactions and radiation studies15,16,17,18,19,20,21,22. Three-electron σ-bonds are most frequently observed in radical cations (Fig. 1a), where they are formed by the interaction of an unpaired electron in a p-orbital of a radical cation with a free p-electron pair from an unoxidized atom, featuring a long weak 2c–3e bond with an antibonding orbital occupied by a single electron and a bond order of 0.5 (hemi-bond). Structure determination and analysis is one of the most useful and direct methods for studies of odd-electron bonds. Although many three-electron σ-bonding systems X∴X and X∴Y (X, Y=He, N, S, P, halogen and so on) have been investigated in the gas phase, solution and solid matrix, and characterized by various spectroscopic techniques in conjunction with theoretical calculations23,24,25,26,27,28,29,30,31, few of them are room-temperature stable because they are either too reactive or dimerize in the solid state. Only three examples have been isolated and structurally characterized by single-crystal X-ray diffraction (Fig. 1b). Gerson et al.32 and Alder et al.33 reported cyclic framework-constrained N∴N three-electron σ-bonds almost 30 years ago. In 1997, Drews and Seppelt34 isolated and structurally characterized the Xe2+ ion containing a Xe∴Xe three-electron σ-bond.

(a) Interaction diagram for the formation of a three-electron σ-bond between a radical cation and a neutral atom. (b) Crystallographically characterized three-electron σ-bonds.
We recently have succeeded in stabilization of a number of interesting radical cations35,36,37,38,39,40 by using the weakly coordinating anion [Al(ORF)4]− (ORF=OC(CF3)3)41. Inspired by those previous results, we herein report the isolation and structure of a Se∴Se three-electron σ-bond, as well as its reversible dimerization. The products were consequently investigated by ultraviolet–visible, electron paramagnetic resonance (EPR), single-crystal X-ray diffraction and superconducting quantum interference device (SQUID) measurements, in conjunction with density functional theory (DFT) calculations.
Results
Isolation of radical cation 1·+
1,8-Dichalcogen naphthalene derivatives have been shown to undergo one- and two-electron oxidations by concentrated H2SO4 to form radical cations and dications42,43,44. The unstable radical cations are suggested to contain a Se∴Se three-electron σ-bond42. Cyclic voltammetry (CV) of 1,8-bis(phenylselenyl)naphthalene (NapSe2Ph2, 1)45 in CH2Cl2 at room temperature with n-Bu4NPF6 as a supporting electrolyte revealed reversible oxidation peaks at oxidation potentials of +0.94 and +1.15 V (Supplementary Fig. 1). In the light of these CV data, 1 was treated with one equiv NO[Al(ORF)4]46 in CH2Cl2 to afford blue radical cation 1·+ in a high yield. The resulting cation is thermally stable under nitrogen atmosphere and can be stored for several months at room temperature.
Crystals suitable for X-ray crystallographic studies were obtained by cooling a solution of 1·+[Al(ORF)4]− in CH2Cl2. Radical cation 1·+is stacked as a dimeric pair (Supplementary Fig. 2) by four Se–Cnaphthalene contacts (3.55 Å, 2 × ; 3.60 Å, 2 × ) that are close to the sum (3.60 Å) of van der Waals radii of selenium and carbon. In the structure of radical cation 1·+, the naphthalene skeleton is essentially coplanar with two selenium atoms (Fig. 2). This is different from neutral 1 where considerable displacement of the selenium atoms from the naphthyl plane is observed (Supplementary Fig. 3). In 1·+, the phenyl rings overlap in a face-to-face offset arrangement with centroid−centroid distance (3.651(1) Å) within the range for typical π−π stacking (3.3−3.8 Å). Both Se−CPh bonds are aligning ‘perpendicular’ to the naphthyl plane. The average C–Se bond lengths are shorter while ∠C–Se–C angles are slightly larger than those in neutral 1. The Se–Se separation (2.942(1) Å) is shorter than that (3.135(2) Å) in 1, but much longer than the Se+–Se+ single-bond length (2.382(2) Å) in 1,5-diselenoniabicyclo[3.3.0]octane dication47. The planarity of the naphthalene-Se–Se backbone, increase of ∠C–Se–C angle and decrease of Se–Se distance compared with neutral 1 indicate the presence of a weak Se–Se bonding interaction in 1·+.

(a,b) 50% ellipsoid drawings of 1·+ with different views. Yellow, carbon; red, selenium; white, hydrogen. Selected bond length (Å) and angle (°): Se1–C17 1.915(2), Se1–C27 1.896(2), Se2–C24 1.923(2), Se2–C33 1.909(2), Se1–Se2 2.942(1), C17–Se1–C27 100.65(10), C24–Se2–C33 100.13(10). (c,d) Spin-density map (c) and SOMO (d) for 1·+ calculated at the UPBE0/SVP level.
To rationalize the experimental results and get further insights into their electronic structures, we carried out DFT calculations for species 1 and 1·+, along with dication 12+. The calculated energy ΔE for the reaction 2 1·+→1+12+ in the solution of CH2Cl2 is +24.1 kcal mol−1, which indicates it is unlikely for the radical cation (1·+) to disproportionate to neutral 1 and dication 12+. X-ray crystal structures of 1 and 1·+ were well reproduced by DFT calculations (Table 1; Supplementary Fig. 4). Consistent with the experimental data, one-electron oxidation of 1 causes a significant decrease of the Se–Se separation by ~0.2 Å, which combined with dominant spin-density distribution (Fig. 2c; Supplementary Table 2) on Se atoms (0.427, 0.421) becomes strong evidence for the formation of a 2c–3e hemi-bond between Se atoms. The calculated Mayer bond order (0.360) for Se−Se further supports the hemi-bond formation. As shown in Fig. 2d, both selenium atoms are main contributors to the singly occupied molecular orbital (SOMO), with Se−Se antibonding character. To check whether the naphthalene scaffold affects the Se−Se bonding, model compounds Me2SeSeMe2·+ and Ph2SeSePh2·+ were also calculated, which afforded linear Se−Se antibonding orbitals with trans configurations (Supplementary Fig. 5). The slight bending of SOMO of 1·+ is due to the constraints imposed by the naphthalene scaffold.
The radical and hemi-bond identification was completed by EPR, nuclear magnetic resonance (NMR), SQUID and ultraviolet–visible measurements. The solution EPR spectrum (Fig. 3a) of 1·+[Al(ORF)4]− at 298 K shows 77Se (spin I=1/2; natural abundance=7.6) satellite peaks, which is attributed to Nap77SePhSePh species. The signal for the Nap77SePh77SePh isotopomer is too weak to observe because of its low concentration. The ratio of main (even-numbered Se isotopes) to satellite (77Se) spectrum intensities is in agreement with the expected value (12.2) and the Aiso (95G) is comparable to that for Me2SeSeMe2·+ (108G) observed in the γ-irradiated sample48. The EPR spectrum of crystalline 1·+[Al(ORF)4]− shows an anisotropic spectrum with gx=2.0011, gy=2.0255 and gz=2.0441 (Supplementary Fig. 6). The giso value (2.0236) is close to that of (Me2SeSeMe2)·+ (giso=2.0344)48 but is significantly smaller than those (2.0639–2.0644) of the diaryl diselenide radical cations (ArSeSeAr)·+ stabilized in pentasil zeolite (Na-ZSM-5)49, where a two-center three-electron π–bond is suggested (Supplementary Fig. 7). The paramagnetic property of radical salt 1·+[Al(ORF)4]− was further confirmed by SQUID measurement (Supplementary Fig. 8) and broad 1H NMR peaks (Supplementary Fig. 9). The ultraviolet–visible spectrum with broad absorption peaks (Fig. 3b) is typical of a three-electron σ-bond and the absorptions are in the range of 370–680 nm for the reported absorptions of S∴S and Se∴Se bonds in the solution50. Judging from the time-dependent DFT calculations of 1·+opt (Supplementary Fig. 10; Supplementary Table 3), absorptions around 580 (ε 21750) and 465 nm (ε 11080) are assigned to overlapped transitions of HOMO(β)→LUMO(β) (I) and HOMO-1(β)→LUMO(β) (II) (580 nm: I 84%, II 12%; 465 nm: I 13%, II 83%).

(a,c) EPR spectra for 1·+[Al(ORF)4]− (a) and 1·+SbF6− (c) in CH2Cl2 (1 × 10−4 M, 298 K). (b,d) Absorption spectra for 1·+[Al(ORF)4]− (b) and 1·+SbF6− (d) in CH2Cl2 (1 × 10−4 M, 298 K).
Dimerization of radical cation 1·+
Further experimental work shows that the formation of Se∴Se bonding is anion dependent. The reaction of 1 with 1 equiv NOSbF6 resulted in a blue solution of 1·+SbF6−. The ultraviolet–visible absorption, EPR spectra (Fig. 3c,d) and broadness of 1H NMR peak (Supplementary Fig. 11) of the reaction solution are similar to those of 1·+[Al(ORF)4]−. Compared with the sharp 19F peak (δ−75.22 p.p.m.)51 in the NMR spectrum of 1·+[Al(ORF)4]−, a very broad 19F NMR peak is observed in that of 1·+SbF6−, suggesting some degree of interaction between ion pairs in the solution of 1·+SbF6− (Supplementary Figs 12 and 13). Concentrating and cooling the blue solution of 1·+SbF6− afforded reddish brown crystals, which were identified as a dimeric complex [1–1]2+(SbF6−)2 by X-ray crystallographic analysis. Redissolving [1–1]2+(SbF6−)2 in CH2Cl2 immediately gave a blue solution with an identical absorption spectrum to 1·+SbF6−. The crystallization and dissolution accompanied with intense colour change demonstrate a reversible process between radical cation 1·+ and dimer [1–1]2+, as shown in Fig. 4a. The solid [1–1]2+(SbF6−)2 exhibits an EPR spectrum (Supplementary Fig. 14), which is similar to that observed for 1·+[Al(ORF)4]−, but a magnetic susceptibility measurement indicates that the brown solid is diamagnetic (Supplementary Fig. 15). The EPR signal of the solid is probably due to trace amounts of radical cation salt 1·+SbF6− trapped in the solid. A similar reversible process with colour change from yellow to red was observed during the oxidation of 1,5-dithiacyclooctane, but has not been identified52.

(a) Reversible dissociation of [1–1]2+. (b) Crystal structure of [1–1]2+ (in [1–1]2+(SbF6−)2) with 50% ellipsoid drawing. Yellow, carbon; red, selenium; white, hydrogen. Selected bond length (Å) and angle (°): Se1–C1 1.916(7), Se1–C11 1.912(8), Se2–C8 1.939(7), Se2–C17 1.931(8), Se1–Se2 2.8815(9), Se2–Se2' 2.9543(13), C1–Se1–C11 101.1(3), C8–Se2–C17 100.2(3), Se1–Se2–Se2' 174.79(4), Se1–Se2–Se2'–Se1' −180.0.
Single-crystal X-ray diffraction showed that [1–1]2+ is composed of two asymmetric subunits symmetrically coupled through a selenium–selenium interaction, giving rise to a linear Se–Se–Se–Se arrangement (Fig. 4b). Compared with the structure of radical cation 1·+, the Se1–C bond distances (Se1–C1, Se1–C11) basically keep unchanged, while the Se2–C bonds (Se2–C17, Se2–C8) are lengthened and the intramolecular Se–Se distance (Se1–Se2) of 2.8815(9) becomes shorter. The Se–Se distance (Se2–Se2′ 2.9543 (13) Å) connecting two subunits is quite long. These Se–Se contacts in [1–1]2+ are comparable to those (2.715(4)–2.929(2) Å) in donor–acceptor complex cations of selone derivatives with TCNQ reported by Bigoli et al.53, Devillanova et al.54, in which the nearly linear arrangement of Se–Se–Se is viewed as a three-center two-electron bond. The unusual dimeric structure of [1–1]2+ resembles the dimer of unstable tellurium-centered (TePiPr2NiPr2PTe)· radical55. It is also worth noting that dialkyl dichalcogen radical cation (MeSe)2·+, containing a two-center three-electron Se–Se π-bond, dimerizes as a rectangular species (MeSe)42+ by a π*–π* interaction with long Se–Se bonds (2.974(1) Å)56 that are comparable to those of [1–1]2+. However, in these cases reversibility was not observed53,54,55,56.
The crystal structure of the dimer [1–1]2+ has been reproduced as a closed-shell singlet by theoretical calculations. Of particular note, the calculated Se2–Se2′ bond lengths (2.9236 Å) is comparable to that (2.9543(13) Å) in the X-ray structure of [1–1]2+. The HOMO clearly shows a σ*–σ* interaction between two radical cation SOMOs (Fig. 5a). Notably Se2–Se2′ bond length of [1–1]2+ is much longer than that (2.6471(6) Å) of the dicationic dimer by irreversible coupling of hypothetical 1,5-selenathiamesocycle radical cation57. The long and weak intermolecular Se–Se bond in [1–1]2+(SbF6−)2 thus accounts for the reversibility of dimerization, and is due to the multicentered nature of the radical SOMO. However, the magnitude of the electronic coupling between the radical cation moieties in the solid state is sufficient to lead to bulk diamagnetism of [1–1]2+ as proved by SQUID measurements. In terms of the valence bond theory, the Se–Se–Se–Se fragment in [1–1]2+ may be viewed as a 4c–6e bonding stabilized by some resonance structures (Fig. 5b)58.

(a) HOMO of [1–1]2+ showing a σ*–σ* interaction between two radical cation SOMOs. (b) Resonance structures.
Discussion
We here have shown that the diselena species (1) modified with a naphthalene scaffold can undergo one-electron oxidation using a large and weakly coordinating anion [Al(ORF)4]−, to afford a room-temperature-stable radical cation (1·+) containing a Se∴Se three-electron σ-bond. When a smaller anion SbF6− is used, a reversible dimerization with phase and marked colour changes is observed: radical cation (1·+) in blue solution but brown diamagnetic dimer ([1–1]2+) in the solid state. The energy ΔE calculated at the (U)PBE0/SVP level for the dimerization 2 1·+→[1–1]2+ is +37.2 and +3.82 kcal mol−1 in the gas phase and CH2Cl2 solution, respectively, indicating it is thermodynamically unfavourable for 1·+ to dimerize in the gas phase or in solution. This is due to the strong electrostatic repulsion of the two adjacent positive charges in [1–1]2+, which is relieved upon dissociation. The unstable dimeric species [1–1]2+ in the gas phase is lattice stabilized by the formation of the salt [1–1]2+(SbF6−)2 in the solid state, because the lattice energy for [1–1]2+(SbF6−)2 is about three times that of 1·+SbF6− assuming ionic radii are similar, as shown by the Kapustiniskii equation (Fig. 6a)41,59,60,61, where zx and zy are the charge of the ions, rx and ry the ionic radii and ν is the number of ions per formula unit (for example, two for X+Y− and three for X2+(Y−)2). As shown in the Born–Fajans–Haber cycle (Fig. 6b), replacement of the anion SbF6− (0.121 nm3) by the larger anion [Al(ORF)4]− (0.736 nm3) leads to a great reduction (less negative) in the lattice energy difference {ΔlattU ([1–1]2+(A−)2)−ΔlattU (21·+A−)}, which would make ΔE(s) more positive and thus favours the formation of singly charged 1·+ over the doubly charged [1–1]2+. Our findings suggest that more examples of three-electron σ-bonds may be stabilized by using naphthalene scaffolds together with large and weakly coordinating anions. Isolation of other examples of those elusive and intriguing three-electron σ-bonds X∴X and X∴Y (X, Y=S, Te, P, As, halogen and so on) is under way.

(a) Kapustiniskii equation for lattice energy estimation. (b) Born–Fajans–Haber cycle rationalizing the formation of the dimer [1–1]2+(A−)2.
Methods
General
All experiments were carried out under a nitrogen atmosphere by using standard Schlenk techniques and a glovebox. Solvents were dried before use. NOSbF6 (Alfa Aesar) was purchased and used upon arrival. 1,8-bis(phenylselanyl) naphthalene (1)45 and NO[Al(ORF)4]46 were synthesized according to the literature methods. CV was performed on an IM6ex electrochemical workstation, with platinum as the working and counter electrodes, Ag/Ag+ as the reference electrode and 0.1 M n-Bu4NPF6 as the supporting electrolyte. EPR spectra were obtained using the Bruker EMX-10/12 variable-temperature apparatus. ultraviolet–visible spectra were recorded on the Lambda 750 spectrometer. Element analyses were performed at Shanghai Institute of Organic Chemistry, the Chinese Academy of Sciences. Magnetic measurements were performed using a Quantum Design MPMS XL-7 SQUID magnetometer at a temperature ranging from 5 to 350 K. The 1H NMR spectra were performed using a Bruker DRX-500 spectrometer in p.p.m. downfield from Me4Si. 19F NMR spectra were performed at ambient temperature on the Bruker DRX-400 spectrometer using CFCl3 as an external reference. X-ray crystal structures were obtained by using a Bruker APEX DUO CCD detector. Crystal data and structure refinement for 1·+[Al(ORF)4]− and [1–1]2+(SbF6−)2 are listed in Supplementary Table 1.
Preparation of 1·+[Al(ORF)4]−
Under anaerobic and anhydrous conditions, a mixture of 1 (0.088 g, 0.2 mmol) and NO[Al(ORF)4](0.199 g, 0.2 mmol) in CH2Cl2 (≈50 ml) was stirred at room temperature for 1 day. The resulting blue solution was then concentrated and stored at around −20 °C for 24 h to afford X-ray-quality crystals of the radical cation salt 1·+[Al(ORF)4]−. Isolated yield: 0.124 g, 44% (crystals); mp 135–137 °C; ultraviolet–visible (CH2Cl2): λmax 465 (ε 11080, shoulder), 580 (ε 21750) nm; analysis (calcd., found for C38H16AlF36O4Se2) C (32.48, 32.82), H (1.15, 1.26).
Preparation of 1·+SbF6− and [1–1]2+(SbF6−)2
Under anaerobic and anhydrous conditions, a mixture of 1 (0.132 g, 0.3 mmol) and NOSbF6(0.080 g, 0.3 mmol) in CH2Cl2 (≈50 ml) was stirred at room temperature for 1 day. The resulting blue solution of 1·+SbF6− was then concentrated and stored at around −20 °C for 24 h to afford X-ray-quality crystals of salt [1–1]2+(SbF6−)2. Isolated yield: 0.103 g, 45% (crystals); mp 153–155 °C; ultraviolet–visible (CH2Cl2): λmax 460 (ε 12190, shoulder), 588 (ε 24320) nm; analysis (calcd., found for C44H32F12Sb2Se4·CH2Cl2): C (37.72, 38.15), H (2.39, 2.57).
Quantum chemical calculations
Calculations were performed with the Gaussian 09 program suite (Supplementary Note 1; Supplementary Reference 2). Geometries were optimized and checked as energy minima by frequency calculations at the (U)PBE0/SVP level of theory. The ultraviolet–visible absorption spectrum was calculated on the optimized geometry using the time-dependent DFT method at the UM06-2X/6-31+G(d) level. To consider solvent (CH2Cl2) effects, a polarized continuum model was adopted in the calculation of the single-point energies involved in the the disproportionation and dimerization, and ultraviolet–visible absorption spectrum. Mayer bond order was calculated at the UPBE0/SVP level with the Multiwfn program62.
Additional information
How to cite this article: Zhang, S. et al. Isolation and reversible dimerization of a selenium–selenium three-electron σ-bond. Nat. Commun. 5:4127 doi: 10.1038/ncomms5127 (2014).
References
Griller, D. & Ingold, K. U. Persistent carbon-centered radicals. Acc. Chem. Res. 9, 13–19 (1976).
Schmittel, M. & Burghart, A. Understanding reactivity patterns of radical cations. Angew. Chem. Int. Ed. 36, 2550–2589 (1997).
Power, P. P. Persistent and stable radicals of the heavier main group elements. Chem. Rev. 103, 789–809 (2003).
Chivers, T. & Konu, J. in:Comprehensive Inorganic Chemistry II: From Elements to Applications, Volume 1: Main-Group Elements, Including Noble Gases ed. Chivers T. 349–373Elsevier (2013).
Zard, S. Z. Radical Reactions in Organic Synthesis Oxford Univ. Press (2003).
Hicks, R. G. Stable Radicals, Fundamentals and Applied Aspects of Odd-Electron Compounds Wiley (2010).
Martin, C. D., Soleilhavoup, M. & Bertrand, G. Carbene-stabilized main group radicals and radical ions. Chem. Sci. 4, 3020–3030 (2013).
Baird, N. C. The three-electron bond. J. Chem. Edu. 54, 291–293 (1977).
Asmus, K. Stabilization of oxidized sulfur centers in organic sulfides. Radical cations and odd-electron sulfur-sulfur bonds. Acc. Chem. Res. 12, 436–442 (1979).
Grützmacher, H. & Breher, F. Odd-electron bonds and biradicals in main group element chemistry. Angew. Chem. Int. Ed. 41, 4006–4011 (2002).
Canac, Y. et al. Isolation of a benzene valence isomer with one-electron phosphorus-phosphorus bonds. Science 279, 2080–2082 (1998).
Kato, T., Gornitzka, H., Schoeller, W. W., Baceiredo, A. & Bertrand, G. Dimerization of a cyclo-1σ4,3σ2,4σ2-triphosphapentadienyl radical: evidence for phosphorus–phosphorus odd-electron bonds. Angew. Chem. Int. Ed. 44, 5497–5500 (2005).
Moret, M., Zhang, L. & Peters, J. C. A polar copper-boron one-electron σ-bond. J. Am. Chem. Soc. 135, 3792–3795 (2013).
Pauling, L. The nature of the chemical bond. II. The one-electron bond and the three-electron bond. J. Am. Chem. Soc. 53, 3225–3237 (1931).
Schöneich, C., Aced, A. & Asmus, K. Mechanism of oxidation of aliphatic thioethers to sulfoxides by hydroxyl radicals. The importance of molecular oxygen. J. Am. Chem. Soc. 115, 11376–11383 (1993).
Miller, B. L., Kuczera, K. & Schöneich, C. One-electron photooxidation of N-methionyl peptides. Mechanism of sulfoxide and azasulfonium diastereomer formation through reaction of sulfide radical cation complexes with oxygen or superoxide. J. Am. Chem. Soc. 120, 3345–3356 (1998).
Schöneich, C., Pogocki, D., Hug, G. L. & Bobrowski, K. Free radical reactions of methionine in peptides: mechanisms relevant to β- amyloid oxidation and alzheimer’s disease. J. Am. Chem. Soc. 125, 13700–13713 (2003).
Majjigapu, K., Majjigapu, J. R. R. & Kutateladze, A. G. Photoamplification and multiple tag release in a linear peptide-based array of dithiane adducts. Angew. Chem. Int. Ed. 46, 6137–6140 (2007).
Musker, W. K. Chemistry of aliphatic thioether cation radicals and dications. Acc. Chem. Res. 13, 200–206 (1980).
Alder, R. W. Medium-ring bicyclic compounds and intrabridgehead chemistry. Acc. Chem. Res. 16, 321–327 (1983).
Abu-Raqabah, A. & Symons, M. C. R. The pyridine-chlorine atom 3-electron-bond intermediate. J. Am. Chem. Soc. 112, 8614–8615 (1990).
Qin, X., Meng, Q. & Williams, F. ESR studies of the thietane and thiirane radical cations in freon matrices. Evidence for ethylene molecule extrusion from the σ* thiirane dimer radical cation [C2H4S-SC2H4·+]. J. Am. Chem. Soc. 109, 6778–6788 (1987).
Drewello, T., Lebrilla, C. B., Asmus, K. & Schwarz, H. Dithia dications from cyclic and acyclic precursors by gas-phase oxidation (‘charge stripping’) of 3e/2c-radical cations. Angew. Chem. Int. Ed. 28, 1275–1276 (1989).
Livant, P. & Illies, A. Estimate of the iodine-iodine two-center three-electron bond energy in [CH3-I-I-CH3]+. J. Am. Chem. Soc. 113, 1510–1513 (1991).
Maity, D. K. Structure, bonding, and spectra of cyclic dithia radical cations: a theoretical study. J. Am. Chem. Soc. 124, 8321–8328 (2002).
Braida, B., Hazebroucq, S. & Hiberty, P. C. Methyl substituent effects in [HnX∴XHn]+ three-electron-bonded radical cations (X=F, O, N, Cl, S, P; n=1–3). An ab initio theoretical study. J. Am. Chem. Soc. 124, 2371–2378 (2002).
Ekern, S., Illies, A., McKee, M. L. & Peschke, M. A novel mechanism for reactions of thiirane with the thiirane radical cation. An experimental and ab initio study. J. Am. Chem. Soc. 115, 12510–12518 (1993).
Deng, Y. et al. A definitive investigation of the gas-phase two-center three-electron bond in [H2S:.SH2]+, [Me2S:.SMe2]+, and [Et2S:.SEt2]+: theory and experiment. J. Am. Chem. Soc. 117, 420–428 (1995).
De Visser, S. P., de Koning, L. J. & Nibbering, N. M. M. Chemical and thermodynamic properties of methyl chloride dimer radical cations in the gas phase. J. Am. Chem. Soc. 120, 1517–1522 (1998).
Nichols, L. S., McKee, M. L. & Illies, A. J. An experimental and theoretical investigation of ion-molecule reactions involving methyl halide radical cations with methyl halides. J. Am. Chem. Soc. 120, 1538–1544 (1998).
Dinnocenzo, J. P. & Banach, T. E. The quinuclidine dimer cation radical. J. Am. Chem. Soc. 110, 971–973 (1988).
Gerson, F., Knöbel, J., Buser, U., Vogel, E. & Zehnder, M. A N-N three-electron σ–bond. Structure of the radical cation N, N′-trimethylene-syn-1, 6:8, 13-diimino[14]annulene as studied by ESR spectroscopy and X-ray crystallographic analysis. J. Am. Chem. Soc. 108, 3781–3783 (1986).
Alder, R. W., Orpen, A. G. & White, J. M. Structures of the radical cation and dication from oxidation of 1,6-diazabicyclo[4.4.4]tetradecane. J. Chem. Soc. Chem. Commun. 1985, 949–951 (1985).
Drews, T. & Seppelt, K. The Xe2+ ion-preparation and structure. Angew. Chem. Int. Ed. 36, 273–274 (1997).
Su, Y. et al. Tuning ground states of bis(triphenylamine) dications: from a closed-shell singlet to a diradicaloid with an excited triplet state. Angew. Chem. Int. Ed. 53, 2857–2861 (2014).
Zheng, X. et al. One-electron oxidation of an organic molecule by B(C6F5)3; isolation and structures of stable non-para-substituted triarylamine cation radical and bis(triarylamine) dication diradicaloid. J. Am. Chem. Soc. 135, 14912–14915 (2013).
Pan, X. et al. Stable tetraaryldiphosphine radical cation and dication. J. Am. Chem. Soc. 135, 5561–5564 (2013).
Pan, X., Chen, X., Li, T., Li, Y. & Wang, X. Isolation and X-ray crystal structures of triarylphosphine radical cations. J. Am. Chem. Soc. 135, 3414–3417 (2013).
Chen, X. et al. Reversible σ-dimerizations of persistent organic radical cations. Angew. Chem. Int. Ed. 52, 589–592 (2013).
Chen, X. et al. From monomers to π-stacks, from nonconductive to conductive. Syntheses, characterization and crystal structures of benzidine radical cations. Chemistry 18, 11828–11836 (2012).
Krossing, I. & Raabe, I. Noncoordinating anions—fact or fiction? A survey of likely candidates. Angew. Chem. Int. Ed. 43, 2066–2090 (2004).
Nenajdenko, V. G., Shevchenko, N. E. & Balenkova, E. S. 1,2-Dications in organic main group systems. Chem. Rev. 103, 229–282 (2003).
Knight, F. R. et al. Synthetic and structural studies of 1,8-chalcogen naphthalene derivatives. Chem. Eur. J. 16, 7503–7516 (2010).
Fujihara, H., Yabe, M., Chiu, J. & Furukawa, N. Peri interaction between selenium atoms in dinaphtho[1,8-b,c]-1,5-diselenocin and 1,8-bis(methylseleno)naphthalene. Tetrahedron Lett. 32, 4345–4348 (1991).
Hayashi, S. & Nakanishi, W. Noncovalent Z…Z (Z=O, S, Se, Te) interactions: how do they operate to control fine structures of 1,8-dichalcogene-substituted naphthalenes? Bull. Chem. Soc. Jpn 81, 1605–1615 (2008).
Decken, A., Jenkins, H. D. B., Nikiforov, G. B. & Passmore, J. The reaction of Li[Al(OR)4] (OR=OC(CF3)2Ph, OC(CF3)3) with NO/NO2 giving NO[Al(OR)4], Li[NO3] and N2O. The synthesis of NO[Al(OR)4] from Li[Al(OR)4] and NO[SbF6] in sulfur dioxide solution. Dalton Trans. 2496–2504 (2004).
Iwasaki, F., Morimoto, M. & Yasui, M. Structure of 1,5-diselenoniabicyclo[3.3.0]octane bis(tetrafluoroborate) acetonitrile solvate. Acta. Cryst. C 47, 1463–1466 (1991).
Nishikida, K. & Williams, F. The ESR spectrum and structure of the dimer radical cation of dimethyl selenide (Me2Se-SeMe2+) in a γ-irradiated single crystal. Chem. Phys. Lett. 34, 302–306 (1975).
Lakkaraju, P. S., Shen, K., Roth, H. D. & Garćia, H. Extended diaryl diselenide radical cations in pentasil zeolite studied by EPR and diffuse reflectance optical spectroscopy. J. Phys. Chem. A 103, 7381–7384 (1999).
Asmus, A. D. Odd-electron bonded sulfur- and selenium-centered radical cations as studied by radiation chemical and complementary methods. Nuleonika 45, 3–10 (2000).
Reisinger, A. et al. Silver–ethene complexes [Ag(η2-C2H4)n][Al(ORF)4] with n=1, 2, 3 (RF=fluorine-substituted group). Chem. Eur. J. 15, 9505–9520 (2009).
Musker, W. K., Wolford, T. L. & Roush, P. B. An investigation of mesocyclic and acyclic dithioether cation radicals and dications. J. Am. Chem. Soc. 100, 6416–6421 (1978).
Bigoli, F. et al. Reaction of 1,2-Bis(2-selenoxo-3-methyl-4-imidazolinyl)ethane(ebis) with TCNQ: crystal structure and characterization of the mixed-valence compound [2(ebis)2+·ebis]·2[(TCNQ)32−]. Inorg. Chem. 35, 5403–5406 (1996).
Devillanova, F. A. et al. Reaction of N,N′-dimethyllimidazolidine-2-selone(4) with TCNQ. Characterisation and X-ray crystal structure of the mixed-valence compound 4·(TCNQ)1.167 . J. Mater. Chem. 10, 1281–1286 (2000).
Chivers, T., Eisler, D. J., Ritch, J. S. & Tuononen, H. M. An unusual ditelluride: synthesis and molecular and electronic structures of the dimer of the tellurium-centered radical [TePiPr2NiPr2PTe]·. Angew. Chem. Int. Ed. 44, 4953–4956 (2005).
Mueller, B., Poleschner, H. & Seppelt, K. Dialkyl dichalcogen cations. Dalton Trans. 4424–4427 (2008).
Evans, D. H. et al. Electrochemical and chemical oxidation of dithia-, diselena-, ditellura-, selenathia-, and tellurathiamesocycles and stability of the oxidized species. J. Org. Chem. 75, 1997–2009 (2010).
Harcourt, R. D. Pauling ‘3-electron bonds’, ‘increased-valence’, and 6-electron 4-center bonding. J. Am. Chem. Soc. 102, 5195–5201 (1980).
Cameron, T. S. et al. Bonding, structure, and energetics of gaseous E82+ and of solid E8(AsF6)2 (E=S, Se). Inorg. Chem. 39, 5614–5631 (2000).
Brownridge, S., Jenkins, H. D. B., Krossing, I., Passmore, J. & Roobottom, H. K. Recent advances in the understanding of the syntheses, structures, bonding and energetics of the homopolyatomic cations of Groups 16 and 17. Coord. Chem. Rev. 197, 397–481 (2000).
Cameron, T. S. et al. Preparation, X-ray crystal structure determination, lattice potential energy, and energetics of formation of the salt S4(AsF6)2·AsF3 containing the lattice-stabilized tetrasulfur [2+] cation. Implications for the understanding of the stability of M42+ and M2+ (M=S, Se, and Te) crystalline salts. Inorg. Chem. 39, 2042–2052 (2000).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Acknowledgements
We thank the National Natural Science Foundation of China (Grants 91122019 and 21171087), the Major State Basic Research Development Program (2013CB922101) and the Natural Science Foundation of Jiangsu Province (Grant BK2011549) for financial support. We are grateful to the High Performance Computing Centre of Nanjing University for providing the IBM Blade cluster system. Part of the computational work has been done on the Sugon TC5000 high performance linux cluster at ITCC.
Author information
Authors and Affiliations
Contributions
S.Z. and Y.Q. performed the chemical experiments, and S.Z. recorded all spectroscopic data. Y.S. and Z.Z. performed the X-ray diffraction studies. Xingyong Wang carried out the calculations. Xinping Wang conceived the project and wrote the paper. All authors discussed the results and manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-15, Supplementary Tables 1-3, Supplementary Note 1 and Supplementary References (PDF 1205 kb)
Rights and permissions
About this article

Cite this article
Zhang, S., Wang, X., Su, Y. et al. Isolation and reversible dimerization of a selenium–selenium three-electron σ-bond. Nat Commun 5, 4127 (2014). https://doi.org/10.1038/ncomms5127
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5127
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
