
Abstract
Malaria is a major public health problem that is actively being addressed in a global eradication campaign. Increased population mobility through international air travel has elevated the risk of re-introducing parasites to elimination areas and dispersing drug-resistant parasites to new regions. A simple genetic marker that quickly and accurately identifies the geographic origin of infections would be a valuable public health tool for locating the source of imported outbreaks. Here we analyse the mitochondrion and apicoplast genomes of 711 Plasmodium falciparum isolates from 14 countries, and find evidence that they are non-recombining and co-inherited. The high degree of linkage produces a panel of relatively few single-nucleotide polymorphisms (SNPs) that is geographically informative. We design a 23-SNP barcode that is highly predictive (~92%) and easily adapted to aid case management in the field and survey parasite migration worldwide.
Similar content being viewed by others

Introduction
Malaria threatens nearly half the world’s population, and the deadliest form caused by Plasmodium falciparum remains a leading cause of childhood mortality worldwide1. As countries move closer to elimination and parasites develop tolerance of artemisinins2,3, understanding the inter-connectedness of parasite populations and tracing the source of imported infections have become top priorities. Genetic markers have proved extremely valuable in the eradication of other diseases (for example, polio4). Analysis of nuclear genome variation in P. falciparum (14 chromosomes, 23 Mbp, 19.1% GC content) has been used to identify candidate artemisinin-resistant loci3, and can be exploited to map the dispersion of parasites worldwide and trace the migration of drug-resistant parasites into new areas. Thus, a universal P. falciparum genotyping tool able to interrogate geographically restricted single-nucleotide polymorphisms (SNPs) would be of great value. Current barcoding approaches5 based on nuclear SNPs are constrained by a lack of geographic specificity and frequent recombination, which disrupts multi-locus SNP associations in each generation. To overcome these limitations, we explored the usefulness of the extra-nuclear genomes of the mitochondrion and apicoplast organelles. We postulated that strict maternal inheritance might exclude recombination and so create a barcode that is stable and geographically informative over time.
The mitochondrion genome (mt) of P. falciparum is a 6-kb concatenated linear sequence, is transmitted in female gametocytes and does not recombine among lineages6,7,8; thus, sequence polymorphism in mt is attractive as a potential barcoding tool. Analysis of global sequence variation in mt has revealed geographic differentiation9,10,11, but the limited numbers of SNPs restrict its capacity to resolve fine-scale population differentiation. Apicoplasts are relict non-photosynthetic plastids found in most protozoan parasites belonging to the phylum Apicomplexa, including all Plasmodium species, and show phylogenetic homology to the chloroplasts of plants and red algae12,13. Although the apicoplast has lost any ancestral photosynthetic ability, it retains a genome encoding lifecycle-specific, essential metabolic and biosynthetic pathways that generate isoprenoids, fatty acids and haem13. As these are distinct from homologous human pathways, the apicoplast is an enticing target for antimalarial drugs13,14,15,16,17. The apicoplast genome (apico) is a 35-kb circular sequence6 and is also maternally inherited. Although polymorphism in apico (29.4 kb annotated core, 30 genes, 13.1% GC content) has not been well characterized, it is potentially greater than that in mt (6 kb, 3 genes, 31.6% GC content) owing to its larger size. To develop a robust mt/apico barcode and improve our understanding of apico evolution, a definitive analysis of apico SNP variation in multiple P. falciparum populations is needed to determine the extent of global diversity and existence of recombination.
Although there is good evidence that chloroplasts and mitochondria are co-inherited in plants18, this is not a hard-and-fast rule in other organisms19. Evidence from the laboratory indicates that mt and apico are co-transmitted during P. falciparum gametocytogenesis7, but evidence from the field is lacking. Here, using sequence data from 711 parasite isolates in 14 countries across four continents, we catalogue 151 mt SNPs and 488 apico SNPs and use them to investigate organelle DNA co-inheritance and geographic differentiation at the population level. We find high linkage disequilibrium (LD) between mt and apico SNPs within populations, providing strong evidence that the organelles are indeed co-transmitted and non-recombining. This finding represents a breakthrough in the genetic barcoding of P. falciparum, as it reveals novel extended haplotypes specific for different geographic settings. Using SNP variation of the combined organelle genome (mt/apico) in an iterative haplotype-based classification analysis, we construct a 23-SNP barcode that identifies the region of sample origin with 92% predictive accuracy.
Results
To the 3D7 reference genome we aligned high-quality raw sequence data from 711 P. falciparum samples in five geographic regions: West Africa (WAF: Burkina Faso, Gambia, Ghana and Mali, N=401), East Africa (EAF: Kenya, Malawi and Uganda, N=98), Southeast Asia (SEA: Cambodia, Thailand and Vietnam, N=164), Oceania (OCE: Papua New Guinea, N=25) and South America (SAM: Colombia and Peru, N=23). The sequence coverages of mt (~1,000-fold) and apico (~100-fold) are ~22-fold and ~two-fold greater than the nuclear genome, respectively (Fig. 1; Supplementary Fig. 1). These fold differences in coverage are consistent with known organelle copy numbers in single P. falciparum parasites20,21. Using all sample alignments, we identified 151 high-quality SNPs in mt (25.3 SNPs per kilobase, 65.6% in coding regions) and 488 in apico (16.6 per kilobase, 77.5% in coding regions) (Supplementary Table 1). Of the 151 SNPs, only 20 (13.2%) were identified previously9,10. Across all samples, 65.4% (418/639) of SNPs were singletons, 92.5% (591/639) were rare (minor allele frequency, MAF <1%), 7.5% (48/639) had a MAF >1% and 2.3% (15/639, 3 mt and 12 apico) were common (MAF >5%) (Supplementary Table 1). Multi-allelic SNPs were identified in both genomes (mt 4.0%, apico 5.1%); 29 were tri-allelic and two were quad-allelic (Supplementary Table 1). Of the multi-allelic SNPs, only the quad-allelic locus mt1692 described previously10 has a combined MAF >5%.

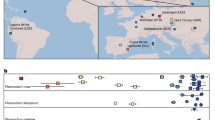
The nucleotide sequence landscape of the densely packed P. falciparum mitochondrion (mt) and apicoplast (apico) genomes. Protein-coding (green) and non-translated RNA (blue) regions in the ‘annotation’ ring are transcribed from either strand (inner, negative strand; outer, positive strand). The 20-fold difference in coverage between the genomes is visible (see also Supplementary Fig. 1). All mutations within mt (151 SNPs, 5,967-bp linear) and apico core (488 SNPs, 29,430-bp circular, excluding an inverted repeat) are shown relative to the P. falciparum 3D7 (version 3.0) reference genome coordinates. SNPs are densely packed throughout, with more non-synonymous (NS) protein-coding changes (red) in apico than in mt. Synonymous, intronic, intra-genic (green) and RNA changes (blue) are also marked. The minor allele frequency (MAF), Fst and barcode SNPs are marked in the outer three rings and are colour coded in the same way (the full catalogue is available online). The 23 barcoding SNPs (5 mt, 4 NS; 18 apico, 9 NS) are marked in the outer ring.
Geographic patterns of diversity were investigated by linear discriminant analysis of the combined mt and apico SNP data, which revealed clustering by geographic origin of samples (Supplementary Fig. 2). To determine the most significant drivers of population differentiation, we analysed only non-rare SNPs. We calculated population differentiation statistic Fst to identify SNPs with inter-regional allele frequency differences, which range from 0 to 1 with higher values signifying greater differentiation22 (Fig. 1). We found substantially lower population differentiation between countries in the same region (mean 28.5 SNPs per region with Fst >0.05) than between the five regions (mean 58 SNPs with Fst >0.05). Forty-nine SNPs have MAF >1% in at least one region (Supplementary Table 2), 17 (34.7%) of which have Fst >0.1 (Supplementary Fig. 3). Of these 17 (4 mt and 13 apico), 14 are located in genes with 8 non-synonymous (NS) changes (Supplementary Fig. 3; Supplementary Table 2). The two SNP loci with highest Fst (~0.76), mt772 (cox3) and apico6762 (orf101), are in perfect LD (r2=1) and differentiate SEA from other regions (MAFs: overall 16.5%, SAM 0%, WAF 0%, EAF 0%, SEA 69.0% and OCE 20.8%). A third SNP with high Fst (~0.88), apico26659 (rpl23), differentiates Africa (WAF and EAF) and SAM from other regions (Supplementary Fig. 3; Supplementary Table 2).
To assess the extent of recombination between SNPs within and between mt and apico, we carried out intra- and inter-region analyses of LD. Using non-rare biallelic markers, there was near-perfect LD between the combined mt and apico SNPs, within and across geographic regions. This is strong evidence that there is no recombination within or between organelles (mean pairwise D′=0.998 for all regions combined, Supplementary Fig. 4), the latter implying potential co-transmission. To investigate this possibility, we used 146 haplotypes (the observed combinations of SNPs in individual parasite isolates) of mt and 271 haplotypes of apico, respectively (Supplementary Table 1). By comparing the joint mt/apico haplotype frequencies (Supplementary Table 3), we found that the dependence between mt and apico was highly significant (χ2=64,921, d.f.=39,566, P<10−16), providing strong evidence of co-inheritance of the two organelles. This genetic evidence confirms the experimentally observed and theoretically predicted processes involved in gametocytogenesis6,7.
The geographical pattern of mt haplotypes was previously interpreted to reflect radiation of P. falciparum out of Africa into SEA and SAM10. Consistent with this interpretation, our analysis of 151 mt SNPs identifies a common haplotype in 30.0% (213/711) of samples, which is represented in four of the five regions: SAM 30.4%, WAF 37.2%, EAF 49.0%, SEA 0% and OCE 36% (Supplementary Fig. 5). Since this compromises geographical assignment, mt haplotypes alone cannot identify the geographic origin of parasite strains. The addition of 488 apico SNPs to generate 290 distinct compound (mt/apico) haplotypes greatly increases the geographic resolution of samples (Supplementary Fig. 5). Nearly all (282/290, 97.2%) compound haplotypes are observed in one region only, and 66.8% of all parasite isolates have a haplotype unique to their region of origin. Six of the eight mt/apico haplotypes observed in multiple regions are most common in Africa (WAF and EAF), consistent with an African origin for this parasite species.
After discovering the existence of regional differentiation, we sought to identify a minimal set of barcoding SNPs diagnostic for the compound mt/apico haplotypes. Using the 221 SNP loci with non-singleton alleles, we applied an iterative haplotype search algorithm that maximized predictive accuracy, while accounting for regional sample size differences and avoiding over-fitting. The minimal barcode comprises 23 SNPs (5 mt, 18 apico, MAF >1% in a single region, 3 tri-allelic), within 18 protein-coding genes (13 NS), four non-translated RNA segments and one inter-genic region (Supplementary Table 2; Fig. 1). The 23 SNPs form only 34 distinct haplotypes (Fig. 2), 26 of which are unique to one region. The core 3D7 haplotype 10 occurs in 14 African isolates (2.8%, 7 WAF and 7 EAF). Haplotypes 14 and 30 occur in three regions and deviate from the core by single mutations.

The 23 SNP loci form 34 distinct haplotypes that help identify a parasite’s geographical origin: South America, SAM; West Africa, WAF; East Africa, EAF; Southeast Asia, SEA; and Oceania, OCE. Most (76.5%, 26/34) haplotypes are unique to a single region. Haplotype 10 corresponds to the 3D7 reference strain, and its mitochondrion (mt) core haplotype is observed in all five regions. Two haplotypes (14 and 30) are seen in three regions. The overall accuracy is 92.1% (655/711; SAM 100%, WAF 94.5%, EAF 68.4%, SEA 98.8% and OCE 96.0%).
The overall predictive accuracy of the minimal barcode is 92.1% (655/711, Supplementary Table 4), compared with 95.1% (676/711) using all 639 mt and apico SNPs, and 82.1% using 24 nuclear SNPs5 (Supplementary Fig. 5). Across all regions except EAF, the predictive accuracy using the barcode is at least 94%. Almost half the discrepancies (24/56) arise from EAF samples being assigned to WAF. The high diversity in EAF samples leads to poor identification using the full and barcoding sets of SNPs, highlighting the need for further characterization of sample genomes from this region. The 23-SNP barcode was validated on sequence data from 81 P. falciparum samples not used in its construction, including five laboratory-adapted clones (3D7, HB3, 7G8, DD2 and GB4 (ref. 23)23), eight samples from travellers returning to London from EAF or WAF24, 154 samples from Africa (Senegal, N=12 (ref. 25)25; Ghana, N=16; Guinea, N=106 (ref. 26)26; Malawi, N=20 (ref. 27)27) and 20 samples from SEA28. The geographic origins of 93.0% (174/187) of isolates were correctly assigned; the origins of eight Malawian (EAF) and five Guinean (WAF) parasites were unassigned as their haplotypes are found in both East and West Africa (haplotypes 8–14, see Fig. 2).
Discussion
Worldwide genetic variation in P. falciparum reflects population history, demography and geographic distance; however, recombination disrupts signals of differentiation in the nuclear genome, and since organelle sequence is non-recombining it can be uniquely informative when tracing patterns of dispersal. Mitochondrial and chloroplast sequences are commonly used in DNA barcodes for animals and plants29 and have been used to explore the origins of humans30 and wine grapevines31. Using genetic variation in the multi-copy mt and apico genomes, we have established a 23-SNP barcode that is geographically informative and robust to the effects of recombination. Rapid sequencing and genotyping technologies can be applied to small amounts of relatively low-grade parasite material, such as that sourced from finger-prick bloodspots. Exploiting uniqueness in the sequences surrounding the informative SNPs supports highly specific identification. The application of this tool has the potential to improve the management of imported cases and reduce the risk of local epidemics resulting from further transmission. Hence it will be a valuable tool for local agencies in programmes of malaria elimination and resistance containment.
The geographic differentiation seen in organelle genomes may also be subject to evolutionary forces in addition to genetic drift and migration. The presence of a core haplotype is consistent with P. falciparum radiation from Africa in the recent past, while sequence analysis using Tajima’s D metric32 supports population expansion in Africa and Asia, and possibly Oceania, and suggests a neutrally mutating population in South America (Supplementary Fig. 6)—all consistent with previous studies of mitochondrial genome diversity10. We explored the possibility that selective forces are also influential. Drug pressure, for example, is exerted regionally through sequential roll-outs of new antimalarial treatments in response to emerging drug resistance. The resulting selective sweeps identified in the nuclear genome have regional dispersal patterns25,33,34,35. In the mitochondrial genome, mutation in codon 268 of cytb occurred in vitro in the presence of atovaquone-proguanil selection11. However, we previously observed no naturally occurring polymorphisms in codons 133, 268 or 280 of this gene25. Since the mitochondrion is a putative target of the antimalarial action of artemisinins33, we looked for association between non-rare mt/apico SNPs and putative artemisinin-resistant loci (chromosome 13 region3 and UBP1 (ref. 36)) but found weak correlation (mean r2=0.00257, maximum r2=0.515). We also considered nuclear SNPs known to be associated with resistance to chloroquine (crt, mdr1, mean r2=0.00454, maximum r2=0.621) and antifolates (dhfr, dhps, mean r2=0.00837, maximum r2=0.371), but again found only weak correlation.
A striking observation is the high proportion of NS changes among coding SNPs in apico (77.8%) compared with mt (31.3%) and the nuclear (61.8%) genome27, which may suggest they are subject to different selective pressures. While all mt genes have low NS to S ratios, indicative of purifying selection and a conserved functional role, apico genes generally have high NS to S ratios indicative of divergence and directional selection37. Drugs may exert selection; the highest NS ratios were in rp8, rps7 and tufA (Supplementary Table 5), the latter encoding a target of the antibiotic thiostrepton and its derivatives38. A more prosaic explanation is nucleotide bias through the unusual apico DNA replication machinery39. To explore this further, we compared NS to S ratios among apico-encoded proteins and 545 nuclear-encoded apicoplast proteins40. The high rate appears to be confined to those apicoplast proteins encoded in apico itself (77.8% NS) rather than the nuclear genome (60.6% NS), thus supporting the DNA replication hypothesis. A similar analysis of mt-encoded proteins and 381 nuclear-encoded mitochondrial proteins41 found NS rates of 55.6% in the nuclear genome and 31.3% in mt. This points to a conservation mechanism that is intrinsic to the mitochondrial sequence. It is also significant that the absence of recombination introduces a constraint on the selective removal of slightly deleterious mutations42, and it is possible that mutations accumulate in sequences linked to genes under strong directional selection. However, multi-copy states of mt and apico within individual parasites may allow deleterious copies to be jettisoned by intracellular selection.
The apicoplast shares evolutionary similarities with the chloroplasts of photosynthetic eukaryotes and the prokaryotic progenitors of all plastids, and is vital to the survival of Plasmodium species13,17. The organelle thus encodes functions absent from vertebrate hosts and presents an enticing target for antimalarial drugs14,15,16,17, including novel applications of known antibiotics and herbicides. By combining these insights with reverse-genetic approaches, it may be possible to identify key proteins and metabolic pathways as new candidate drug targets15 and to anticipate their effectiveness in geographically distinct parasite populations.
An ability to determine the geographic origin of P. falciparum isolates potentially has enormous practical utility in containing drug resistance and eliminating malaria. One potential limitation of the mt/apico barcode in its current form is the lack of representation of the Indian sub-continent, Central America, southern Africa and the Caribbean, owing to the scarcity of sequence data from these regions. In addition, there is a need to sample more intensively from EAF, a region of high genetic diversity, high migration and poor predictive ability. Once these data gaps are filled, the barcode can be re-calibrated to maximize its accuracy in assigning sample origin. The 23 SNPs can be modified in light of new sequence information to improve barcode specificity, especially for discriminating malaria importation from one or two known regions, in which case a minimal set can be applied. Adding genomic data from P. vivax and P. knowlesi should help broaden the scope of the barcode for pan-Plasmodium applications. Incorporating antimalarial drug-resistant loci3 will further enhance the usefulness of the barcode as an important tool for malaria control and elimination activities worldwide.
The demonstration that mt and apico sequences are non-recombining creates a new genotyping tool that is robust to the diluting effects of recombination. Global movement of parasites threatens elimination and treatment efficacy. By mapping global patterns of organellar genome polymorphism, we will gain new insights into the extent to which P. falciparum populations worldwide are inter-connected by international malaria migration.
Methods
Sequence data alignment and variant detection
Raw deep-sequence data (minimum read length 54 base pairs (bp)) were available from P. falciparum isolates sourced from Burkina Faso and Mali27,28,43,44,45, Ghana43, Gambia27,43,46, Guinea26, Kenya28,36, Malawi27, Thailand and Cambodia27,28,43,45, Colombia47 and Vietnam43, as well as laboratory-adapted clones (DD2, HB3, 7G8 and GB4 (ref. 23)23) (also see ref. 27). mt sequence data for 101 samples (SAM 26, WAF 20, EAF 8, SEA 30, OCE 11 and other 6) were also available10.
All sequences were mapped uniquely onto the 3D7 reference genome (14 chromosomes, 23 Mb; mitochondrion, 6 kb; apicoplast core, 30 kb; version 3.0) using smalt alignment software (www.sanger.ac.uk/resources/software/smalt) with default settings within an established pipeline24,27. The resulting alignments enabled the identification of high-quality (Q30) SNPs and small insertions/deletions (indels) using SAMtools and BCF/VCF tools (samtools.sourceforge.net). Genotypes were called using coverage as described24,27, where a minimum of 10-reads support was required to call an allele.
Population genetics and statistical analysis
A linear discriminant analysis was performed to cluster parasite isolates on the basis of genetic information, specifically using pairwise identity by state based on SNP allele differences. SNPs identified in the nuclear genome (~600 K SNPs, http://pathogenseq.lshtm.ac.uk/plasmoview27) were used in a principal component analysis to identify potential geographical outliers. Analyses of allele frequency distributions were performed using within-population Tajima’s D indices32 and between-population Fst22. Negative Tajima’s D values signify an excess of low-frequency polymorphisms relative to expectation, indicating population size expansion (for example, after a bottleneck or a selective sweep) and/or purifying selection. Positive Tajima’s D values signify low levels of low- and high-frequency polymorphisms, indicating a decrease in population size and/or balancing selection. Fst metric values range from 0 (equivalent allele frequencies across populations) to 1 (complete differentiation for at least one population). The Ka/Ks ratio was calculated as an indicator of selective pressure acting on a protein-coding gene (Supplementary Table 5). It is the ratio of the number of NS substitutions per NS site (Ka) to the number of synonymous (S) substitutions per S site (Ks)48. Increasing values of Ka/Ks from 1 imply positive selection, while values decreasing from 1 imply purifying selection. LD was assessed using pairwise D' and r2 methods49. The barcode was constructed using an iterative SNP algorithm that considers the classification of regions using haplotypes, attempting to maximize predictive accuracy (weighted or unweighted by regional sample size) without over-fitting. The search strategy led to a more accurate barcode when compared with traditional SNP (not haplotype)-based approaches, including the incremental addition of SNPs with highest MAF or Fst, as well as classification and regression tree50 and random forest algorithms51 (Supplementary Fig. 7). All statistical analyses were performed using R software (www.r-project.org).
Additional information
How to cite this article: Preston, M.D. et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat. Commun. 5:4052 doi: 10.1038/ncomms5052 (2014).
References
WHO. World Malaria Report 2012, WHO (available at http://www.who.int/ malaria/publications/world_malaria_report_2012/report/en/index.html) (2012).
Phyo, A. P. et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 379, 1960–1966 (2012).
Ariey, F. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55 (2014).
Luo, H. M. et al. Identification and control of a poliomyelitis outbreak in Xinjiang, China. N. Engl. J. Med. 369, 1981–1990 (2013).
Daniels, R. et al. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar. J. 7, 223 (2008).
Creasey, A. et al. Maternal inheritance of extrachromosomal DNA in malaria parasites. Mol. Biochem. Parasitol. 65, 95–98 (1994).
Okamoto, N., Spurck, T. P., Goodman, C. D. & McFadden, G. I. Apicoplast and mitochondrion in gametocytogenesis of Plasmodium falciparum. Eukaryot. Cell 8, 128–132 (2008).
Baker, D. A. Malaria gametocytogenesis. Mol. Biochem. Parasitol. 172, 57–65 (2010).
Conway, D. J. et al. Origin of Plasmodium falciparum malaria is traced by mitochondrial DNA. Mol. Biochem. Parasitol. 111, 163–171 (2000).
Joy, D. A. et al. Early origin and recent expansion of Plasmodium falciparum. Science 300, 318–321 (2003).
Musset, L., Le Bras, J. & Clain, J. Parallel evolution of adaptive mutations in Plasmodium falciparum mitochondrial DNA during atovaquone-proguanil treatment. Mol. Biol. Evol. 24, 1582–1585 (2007).
Wilson, R. J. et al. Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J. Mol. Biol. 261, 155–172 (1996).
Lim, L. & McFadden, G. I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 749–763 (2010).
Fichera, M. E. & Roos, D. S. A plastid organelle as a drug target in apicomplexan parasites. Nature 390, 407–409 (1997).
Ralph, S. A., D’Ombrain, M. C. & McFadden, G. I. The apicoplast as an antimalarial drug target. Drug Resist. Updat. 4, 145–151 (2001).
Bispo N. A., Culleton R., Silva L. A., Cravo P., Costa F. T. M. A systematic in silico search for target similarity identifies several approved drugs with potential activity against the plasmodium falciparum apicoplast. PLoS ONE 8, e59288 (2013).
Friesen J, J. et al. Natural immunization against malaria: causal prophylaxis with antibiotics. Sci. Transl. Med. 2, 40ra49 (2010).
Olson, M. S. & McCauley, D. E. Linkage disequilibrium and phylogenetic congruence between chloroplast and mitochondrial haplotypes in Silene vulgaris. Proc. R. Soc. Lond. B Biol. Sci. 267, 1801–1808 (2000).
Thyssen, G., Svab, Z. & Maliga, P. Exceptional inheritance of plastids via pollen in Nicotiana sylvestris with no detectable paternal mitochondrial DNA in the progeny. Plant J. 72, 84–88 (2012).
Waller, R. F. & McFadden, G. I. The apicoplast: a review of the derived plastid of apicomplexan parasites. Curr. Issues Mol. Biol. 7, 57–80 (2005).
Williamson, D. H. et al. The plastid DNA of the malaria parasite Plasmodium falciparum is replicated by two mechanisms. Mol. Microbiol. 45, 533–542 (2002).
Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat. Rev. Genet. 10, 639–650 (2009).
Sepúlveda, N. et al. A Poisson hierarchical modelling approach to detecting copy number variation in sequence coverage data. BMC Genomics 14, 128 (2013).
Robinson, T. et al. Drug-resistant genotypes and multi-clonality in Plasmodium falciparum analysed by direct genome sequencing from peripheral blood of malaria patients. PLoS ONE 6, e23204 (2011).
Park, D. J. et al. Sequence-based association and selection scans identify drug resistance loci in the Plasmodium falciparum malaria parasite. Proc. Natl Acad. Sci. USA 109, 13052–13057 (2012).
Mobegi, V. A. et al. Genome-wide analysis of selection on the malaria parasite Plasmodium falciparum in West African populations of differing infection endemicity. Mol. Biol. Evol (epub ahead of print) (2014).
Preston, M. D. et al. PlasmoView: a web-based resource to visualize global Plasmodium falciparum genomic variation. J. Infect. Dis 209, 1808–1815 (2014).
Manske, M. et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379 (2012).
Pečnikar, F. Ž. & Buzan, E. V. 20 years since the introduction of DNA barcoding: from theory to application. J. Appl. Genet. 55, 43–52 (2014).
Cann, R. L. et al. Mitochondrial DNA and human evolution. Nature 325, 31–36 (1987).
Imazio, S. et al. Chloroplast microsatellites to investigate the origin of grapevine. Genet. Resour. Crop Evol. 53, 1003–1011 (2006).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Wang, J. et al. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS One 5, e9582 (2010).
Sidhu, A. B. et al. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis. 194, 528–535 (2006).
Wootton, J. C. et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418, 320–323 (2002).
Borrmann S, S. et al. Genome-wide screen identifies new candidate genes associated with artemisinin susceptibility in Plasmodium falciparum in Kenya. Sci. Rep. 3, 3318 (2013).
Arisue, N. et al. The Plasmodium apicoplast genome: conserved structure and close relationship of P. ovale to rodent malaria parasites. Mol. Biol. Evol. 29, 2095–2099 (2012).
Dahl, E. L. & Rosenthal, P. J. Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol. 24, 279–284 (2008).
Seow, F. et al. The plastidic DNA replication enzyme complex of Plasmodium falciparum. Mol. Biochem. Parasitol. 141, 145–153 (2005).
Foth, B. J. et al. Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science 299, 705–708 (2003).
Bender, A. et al. Properties and prediction of mitochondrial transit peptides from Plasmodium falciparum. Mol. Biochem. Parasitol. 132, 59–66 (2003).
Hershberg, R. et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6, e311 (2008).
Miotto, O. et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 45, 648–655 (2013).
Auburn, S. et al. Characterization of within-host Plasmodium falciparum diversity using next-generation sequence data. PLoS One 7, e32891 (2012).
Preston, M. D. et al. VarB: a variation browsing and analysis tool for variants derived from next-generation sequencing data. Bioinformatics 28, 2983–2985 (2013).
Amambua-Ngwa, A. et al. SNP genotyping identifies new signatures of selection in a deep sample of West African Plasmodium falciparum malaria parasites. Mol. Biol. Evol. 29, 3249–3253 (2012).
Echeverri-Garcia, D. F. Population Genetics and Polymorphisms in Plasmodium Falciparum Parasites from Colombian Pacific Region before the Introduction of Artemisinine Compounds University of Purdue (2013).
Hurst., L. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486–489 (2002).
Mueller, J. C. Linkage disequilibrium for different scales and applications. Brief Bioinform. 5, 355–364 (2004).
Breiman, L. et al. Classification and Regression Trees Wadsworth & Brooks/Cole Advanced Books & Software (1984).
Liaw, A. & Wiener, M. Classification and regression by randomforest. R News 2, 18–22 (2002).
Acknowledgements
M.D.P. and T.G.C. received funding from the UK Medical Research Council. H.O. is supported by the Malaria Capacity Development Consortium, which is funded by The Wellcome Trust (WT084289MA). C.J.S. is supported by Public Health England. We wish to thank Scott Jackson, Catherine Hill, Tim Anderson and the malaria research team at CIDEIM for providing Colombian parasite samples, and the investigators who placed their sequencing data into the public domain. We wish to thank the Malaria Programs at the Wellcome Trust Sanger and Broad Institutes for putting raw sequence data into the public domain. This work was supported in part by the Intramural Research Program of the NIH, NIAID.
Author information
Authors and Affiliations
Contributions
S.C. and T.G.C. conceived the project. S.A.A., D.F.E., H.O., A.A.-N., L.B.S., D.J.C., S.B., P.M., I.Z., J.-B.O., A.A.D., O.K.D., F.N., A.P., T.B., C.J.D., R.M.F. and C.J.S. contributed to the construction of data. M.D.P., S.C. and T.G.C. analysed the data. C.R. and T.G.C. jointly supervised the research. M.D.P., R.M.F., C.R. and T.G.C. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-7 and Supplementary Tables 1-5 (PDF 1012 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Preston, M., Campino, S., Assefa, S. et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun 5, 4052 (2014). https://doi.org/10.1038/ncomms5052
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5052
This article is cited by
-
Anti-malarial resistance in Mozambique: Absence of Plasmodium falciparum Kelch 13 (K13) propeller domain polymorphisms associated with resistance to artemisinins
Malaria Journal (2023)
-
Rapid profiling of Plasmodium parasites from genome sequences to assist malaria control
Genome Medicine (2023)
-
Synthesis and antimalarial activity of amide and ester conjugates of siderophores and ozonides
BioMetals (2023)
-
5WBF: a low-cost and straightforward whole blood filtration method suitable for whole-genome sequencing of Plasmodium falciparum clinical isolates
Malaria Journal (2022)
-
Gene Coverage Count and Classification (GC3): a locus sequence coverage assessment tool using short-read whole genome sequencing data, and its application to identify and classify histidine-rich protein 2 and 3 deletions in Plasmodium falciparum
Malaria Journal (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
