
Abstract
Metal-organic frameworks (MOFs) based on zirconium phosphonates exhibit superior chemical stability suitable for applications under harsh conditions. These compounds mostly exist as poorly crystallized precipitates, and precise structural information has therefore remained elusive. Furthermore, a zero-dimensional zirconium phosphonate cluster acting as secondary building unit has been lacking, leading to poor designability in this system. Herein, we overcome these challenges and obtain single crystals of three zirconium phosphonates that are suitable for structural analysis. These compounds are built by previously unknown isolated zirconium phosphonate clusters and exhibit combined high porosity and ultrastability even in fuming acids. SZ-2 possesses the largest void volume recorded in zirconium phosphonates and SZ-3 represents the most porous crystalline zirconium phosphonate and the only porous MOF material reported to survive in aqua regia. SZ-2 and SZ-3 can effectively remove uranyl ions from aqueous solutions over a wide pH range, and we have elucidated the removal mechanism.
Similar content being viewed by others
Introduction
The long-term stability of metal-organic framework (MOF) compounds remains a challenge. During the past decade, significant amounts of works have been devoted to designing and synthesizing MOFs that are resistant to structural degradation induced by moisture, acids/bases, oxidants/reductants and radiation, critical features for many practical applications1,2,3,4,5. To achieve these goals, Pearson hard and soft acid and base theory was followed6,7. For instance, hydrolytically stable zeolitic imidazolate framework materials can be assembled by combining soft low-valence metal cations and soft N-donor ligands, with some examples that can even survive in concentrated/saturated base solutions8,9. For systems with carboxylate ligands containing hard oxo donors, hard Lewis acids, that is, metal cations with high-charge density, which are typically represented by tetravalent zirconium, have been intensively studied for the construction of stable MOFs6,10. Although debates on the hydrolytic stability of some zirconium based MOFs remain11,12, examples exhibiting decent stabilities under extremely acidic conditions (for example, concentrated HCl solutions) have been shown13,14,15,16. However, there has been lacking a single example of MOF compound that can survive all fuming acids including nitric acid, aqua regia and oleum solutions that are often required for a variety of chemical processes such as used nuclear fuel reprocessing (for example, 8 M HNO3).
Compared to zirconium carboxylate MOFs, tetravalent zirconium phosphonates are proposed to possess elevated stabilities owing to the higher pKa values of the phosphonate ligands17. Indeed, the zirconium phosphonates reported in literatures are a class of versatile materials with promising applications such as catalyst supporters, heavy metal sorbents, as well as proton-conducting materials18,19,20,21,22. However, there are two key hurdles for the further development of zirconium phosphonate MOFs. First, owing to the extraordinarily strong affinity between Zr4+ and phosphonate ligands, the poor solubility of zirconium phosphonates often leads to rapid precipitation products with poor crystallinity or even completely amorphous solids known as unconventional MOFs, pioneered by Clearfield and Alberti23. Although the structures of some of these materials were partially solved by powder X-ray diffraction data, a single-crystal structure of a zirconium phosphonate MOF material with precise structural information has remained elusive. Second, most reported zirconium phosphonate MOFs are based on inorganic zirconium phosphate 1D chains or 2D sheets and there has been lacking a zero-dimensional zirconium phosphate cluster that can act as the secondary building unit (SBU), leading to poor designability of the zirconium phosphonate MOF system24. By contrast, Zr6 clusters are found in several benchmark zirconium carboxylate MOFs UiO-66, PCN-222, MOF-525 and other compounds13,25,26,27,28,29, while NU-1000 contains a Zr8 cluster as the SBU30. As another consequence, zirconium phosphonates with 3D framework structures are quite rare24,31,32,33. Among these, only UPG-1, whose structure was solved by powder X-ray diffraction recently, shows permanent porosity33.
To overcome these issues, we pursued two strategies in our synthetic efforts: first, a sterically demanding spacer molecule was adopted to prevent the formation of a 2D structure stemming from the stacking of the layers that was recently reported by Taddei et al. in Zrbtbp constructed using a triangular planar ligand, 1,3,5-tris(4-phosphonophenyl)benzene (H6btbp)34. Alternatively, tetrakis[4-(dihyroxyphosphoryl)phenyl]methane (TppmH8) and 1,3,5,7-tetrakis(4-phosphonophenyl)adamantine (TppaH8) with coordination-available phosphonate groups located at four tetrahedral vertexes as the linker was selected to more effectively prevent the formation of the undesired, densely packed structures. Second, ionothermal synthesis was used to avoid the issue of hydrolysis/solvation of Zr4+ in the traditional hydrothermal/solvothermal reactions and to slow down the crystallization kinetics and thereby promote the growth of large single crystals. Indeed, the ionothermal reaction of ZrCl4 with TppmH8 or TppaH8 in the ionic liquid N-butyl-N-methylpyrrolidinium bromide ([C4mpyr][Br]) and N-ethylpyridinium bromide ([C2py][Br]) in the presence of HF afforded three highly crystalline three-dimensional (3D) microporous Zirconium(IV) phosphonates with crystal sizes that are sufficiently large for X-ray analysis. All these compounds are built based on zero-dimensional zirconium phosphate cluster as the SBU, initially opening the way for the reticular design and synthesis in the zirconium phosphonate system. In addition, these compounds exhibit ultrastabilities in acid solutions especially in fuming acids. Furthermore, two of these compounds possess high permanent porosities and excellent uranium uptake capabilities, while one compound is the most porous crystalline zirconium phosphonate reported up to date and is the only porous MOF that survives in aqua regia.
Results
Structure and characterizations of SZ-1
[C4mpyr][Zr2.5(TppmH3)F6]·2.5H2O (Soochow University Zirconium Phosphonate No. 1, SZ-1) crystallizes in a centrosymmetric orthorhombic space group Pbca. Zr4+ ions are located in two coordination spheres of ZrO6 and ZrO3F3, which are surrounded by six and three bridging CPO3 groups from the Tppm ligands, respectively, as guided by strategy 1. ZrO6 is bridged to four ZrO3F3 polyhedra through six phosphonate groups via corner-sharing (Supplementary Tables 1 and 2). Two pairs of ZrO3F3 are further bridged by one CPO3 group each to give a previously unknown eight-connected Zr5P8 cluster (Fig. 1a,b, Supplementary Fig. 11 and Supplementary Table 5) containing no oxide/hydroxide, consistent with the goal of strategy 2. The Zr5P8 clusters are finally linked by the organic moieties of the Tppm ligands, resulting in a 3D anionic framework structure with [C4mpyr]+ cations filled in the pores (Fig. 1c,d). The position of [C4mpyr]+ can be perfectly determined, suggesting a strong interaction between [C4mpyr]+ and the anionic framework. There are two types of interconnected channels in the SZ-1 structure. The first is a continuous channel along the [010] direction, with window dimensions of ∼5.4 × 5.8 Å, and the second, smaller aperture is along the [100] direction. The total void volume of SZ-1 estimated using PLATON is 27.7%, which is smaller than that of the first microporous compound UPG-1 with a void volume of 32.8% (ref. 33). The SBU of Zr5P8 in SZ-1 is substantially different from the inorganic chains constructed by ZrO6 and phosphonate groups in UPG-1, resulting in a unique {410.614.84}{45.6}2 alb topology containing a Zr5P8 cluster as an eight-connected node and the Tppm ligand as a four-connected node (Fig. 1e).

The Tppm ligand (a) is connected to four 8-connected Zr5P8 clusters (b) to generate a 3D network with a sieve-like layered structure in the bc plane (c–e). Colour scheme: Zr, green; P, purple; C, grey.
The porosities of SZ-1 were investigated by sorption isotherms of N2 and CO2 (Supplementary Fig. 16). The results show that SZ-1 exhibits moderate porosity with a N2 Brunauer–Emmett–Teller (BET) and Langmuir surface area of 10.2 and 16.5 m2 g−1, respectively, likely because of the difficulty of exchanging [C4mpyr]+. The fuming acid solutions with large excess of protons were used to exchange the [C4mpyr]+, but this was unsuccessful as the surface area did not change significantly. Nevertheless, this observation provides a direct evidence for the ultrastability in fuming acid as discussed below.
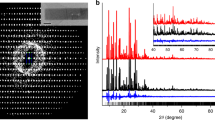
The thermal behaviour of SZ-1 was evaluated by thermogravimetric analysis under nitrogen flow, revealing that SZ-1 is stable up to 300 °C, as further demonstrated by temperature-dependent X-ray diffraction measurements (Supplementary Figs 17 and 18). As one of the most important features, the hydrolytic stabilities of SZ-1 were tested in aqueous solutions. As expected, the crystallinity of the compounds was completely preserved in boiling water and aqueous solutions over a wide pH range (1–11) (Fig. 2a). More impressively, SZ-1 can survive in a variety of fuming acids including 12.2 M HCl, 14.9 M HNO3, aqua regia and oleum (that is, 17.8 M H2SO4) solutions (Fig. 2b). Furthermore, this decent stability in fuming acids was further checked by solid sample mass maintain test confirming that SZ-1 is completely insoluble in these solutions. Furthermore, the crystal structures can be well refined using the diffraction data collected on the oleum/aqua regia-soaked SZ-1 crystals, which are identical with the original structure (Supplementary Fig. 20 and Table 6). This finding is significant since no MOF material has yet been reported to survive in all these fuming acids16. By contrast, BUT-12, 13 and PCN-222 have been shown to survive in concentrated HCl solutions13,14; MOF-808 maintains its crystallinity in 0.1 M H2SO4 solution27; MOF-525 and MOF-545 are stable in water/acetic acid (50/50, vol) solution for one day26; PCN-225 and PCN-230 show excellent stabilities in aqueous solutions over a wide pH range (0–12)3,15; while UPG-1 was shown to be stable under 0.1 M HCl solution at 120 °C for three days33.

Powder X-ray diffraction spectra for compound SZ-1 after soaking in various solutions (a) and in fuming acids (b).
Structure and characterizations of SZ-2
Although the stability of SZ-1 is quite promising, the porosity is still limited, which needs enhancement for further applications. By using more amount of ionic liquids aiming at the expansion of pores, another microporous negative charged 3D framework compound, [C4mpyr]2[Zr1.5(Tppm)0.5F4·6H2O] (SZ-2), was obtained as large single crystals (Supplementary Fig. 13). The synthesis of pure SZ-2 material is found to be very challenging as it often co-crystallizes with SZ-1 under a variety of reaction conditions. To resolve this issue, we introduced a unique technique of slow diffusion into the ionothermal reaction to further slow down the crystallization kinetics (as shown in Supplementary Fig. 10), which allowed us to successfully obtain the pure SZ-2 product as confirmed by the powder X-ray diffraction data. SZ-2 crystallizes in a centrosymmetric tetragonal space group I4/m and is much more porous than SZ-1, as calculated by PLATON. ZrO3F3 and ZrO4F2 moieties are found in SZ-2 and are coordinated by three and four Tppm ligands, respectively (Fig. 3a, Supplementary Tables 1 and 3). Two ZrO3F3 and one ZrO4F2 polyhedra are bridged by four bridging CPO3 groups, resulting in a unique four-connected Zr3P4 cluster (Fig. 3b), and a 3D microporous framework structure is achieved through the linkage of the Tppm ligands with highly disordered C4mpyr+ cations filled in the 3D channels (Fig. 3c,d). Notably, the special arrangement of coordination of Zr3P4 cluster with the organic moiety results in a significant distortion of the central carbon atom coordination in the Tppm ligand deviating from the ideal tetrahedral configuration. Further inspection of the structure reveals that this material should be a good candidate for ion-exchange applications. Three types of apertures are found along the [001] direction, with sizes of 12.86 × 12.86, 11.92 × 11.92 Å, and 3.64 × 1.07 Å (after subtraction of vdW radii of C atoms). The window size for channels along the [100] and [010] directions are close to 1 nm in diameter. The total void volume of SZ-2 is 68.2%, which is the largest reported for a zirconium phosphonate MOFs and is significantly larger than those observed previously. Topological analysis shows that Zr3P4 cluster serves as a four-connected node and the Tppm ligand as a 4-connected node, further leading to a 4-c net with atn topology and a topological point symbol of {42.63.8} (Fig. 3e).

The Tppm ligand (a) is connected to four 4-connected Zr3P4 clusters (b) to generate a 3D network with three types of windows of different sizes along the [001] direction (c–e).
N2 adsorption of SZ-2 shows a significantly elevated surface area of 225 (BET) and 375 (Langmuir) m2 g−1, respectively (Supplementary Fig. 16), compared to that of SZ-1. These values are still lower than those calculated based on the void volume of SZ-2, likely due to the charge-balancing cations in the pore and/or partial framework collapse during the measurement. In comparison, the Langmuir and BET surface area of the neutral framework UPG-1 is 410 and 514 m2 g−1, respectively, as measured using both carbon dioxide and n-butane, while the N2 adsorption measurement did not yield a noticeable uptake value33. The thermal behaviour of SZ-2 was also measured, showing that SZ-2 is stable up to ca. 300 °C (Supplementary Fig. 17). The hydrolytic stabilities tests of SZ-2 reveal that the crystallinity of SZ-2 was completely preserved in boiling water and aqueous solutions over a wide pH range (1–11) (Fig. 4). However, SZ-2 is not able to survive fuming acids, which may be a consequence of the highly distorted central carbon atom coordination in the Tppm ligand.

Powder X-ray diffraction spectra for compound SZ-2 after soaking in various solutions.
Structure and characterizations of SZ-3
Although SZ-1 and SZ-2 exhibit advantages in terms of ultrastability in fuming acids and high porosity respectively, these features do not yet combined in one compound. To solve this issue, we switched ligand from Tppm to Tppa with the change of ligand central portion from a single carbon atom to a much larger adamantane, aiming at reducing the effect of ligand distortion. Indeed, the key compound [C2py]2[Zr3.5(TppaH)F9]·6.5H2O (SZ-3) that is featured with both ultrastability in fuming acids and high porosity was successfully obtained. SZ-3 crystallizes in a monoclinic space group P2(1)/n (Supplementary Tables 1 and 4). Zr4+ ions in SZ-3 are also located in two coordination spheres of ZrO6 and ZrO3F3, which are surrounded by six and three bridging CPO3 groups from the Tppa ligands, respectively (Fig. 5a). Six ZrO3F3 polyhedra are connected to one ZrO6 through six corner-sharing phosphonate groups, giving a sandglass shaped structure terminated by a phosphonate bridging three ZrO3F3 polyhedra. This further gives rise to another previously unknown eight-connected Zr7P8 cluster (Fig. 5b). The Zr7P8 clusters are linked by the organic moieties of the Tppa ligands, resulting in a new anionic 3D framework structure with disordered [C2py]+ cations filled in the pores (Fig. 5c,d). The total void volume of SZ-3 is 46.5%, which is the second largest among all crystalline zirconium phosphonates with known crystal structures, only smaller than SZ-2 in this work. Although the SBU of Zr7P8 in SZ-3 is substantially different from that in SZ-1, the {410.614.84}{45.6}2 alb-4,8-Pbcn topology is the same with that of SZ-1, containing a Zr7P8 cluster as an eight-connected node and the Tppa ligand as a four-connected node (Fig. 5e).

The Tppa ligand (a) is connected to four 4-connected Zr7P8 clusters (b) to generate a 3D network with hybrid porous layers in ac plane (c), which are linked by the organic moieties along b axis (d,e).
The surface area of SZ-3 is evaluated by N2 adsorption, showing a significantly elevated surface area of 572 (BET) and 695 (Langmuir) m2 g−1, respectively (Supplementary Fig. 16), which is noticeably larger than all other crystalline zirconium phosphonate compounds reported up to date. The thermal behaviour of SZ-3 was also measured, showing that SZ-3 is stable up to ca. 210 °C (Supplementary Fig. 17). The hydrolytic stabilities tests of SZ-3 reveal that the crystallinity of SZ-3 was completely preserved not only in aqueous solutions from pH 0 to 11 (Fig. 6a), but also in fuming acids including aqua regia, concentrated HCl, and concentrated HNO3, making SZ-3 currently the only porous MOF material that is reproted to be able to survive aqua regia (Fig. 6b). The N2 adsorption of SZ-3 sample soaked in aqua regia was also tested, giving BET and Langmuir surface area of 594 and 732 m2 g−1, respectively, which are even slightly higher than those of the original sample (Supplementary Fig. 19).

Powder X-ray diffraction spectra for compound SZ-3 after soaking in various solutions (a) and in fuming acids (b).
Uranyl uptake experiments and mechanism elucidation
Inorganic zirconium phosphates and other materials have been intensively investigated as ion-exchange materials for the removal of heavy metal contamination, especially uranium and the fission product caesium, for spent nuclear fuel partitioning and contamination remediation21,35,36,37,38,39,40,41,42. With their significantly improved porosity and surface area, all these three zirconium phosphonate MOFs could be promising candidates for these applications. The uranyl(VI) ion uptake results at various pH show that SZ-2 is able to almost completely remove the uranyl ions over a wide pH range from 3 to 7, while SZ-1 does not exhibit a noticeable ion-exchange capability (Supplementary Fig.21). Common ion-exchange materials, such as inorganic zirconium phosphates often lose their uranium-uptake ability below pH 2 (refs 34, 35, 36, 37, 38, 39, 40, 41). Remarkably, the uranium removal percentage can reach 62.4% using SZ-2 at pH 1.0 (Fig. 7a). This rarely observed result is likely attributed to the combined advantages of both enhanced stability and elevated porosity. The sorption kinetics of uranyl(VI) ion with SZ-2 was also evaluated. At pH 4.5, 72.3% of uranyl ions was removed within only 5 min and the sorption equilibrium with ca. 100% removal ratio was achieved within 3 h (Supplementary Fig. 22). At pH 1.0, the sorption equilibrium can be reached within 5 h, which is typical for ion-exchange materials. Sorption isotherm data fitted by the Langmuir model gives the saturated uranium sorption capacity of 58.18 mg g−1 for SZ-2, which is quite promising compared to the zirconium phosphate based sorbent materials (Fig. 7b). The uranyl sorption selectivity by SZ-2 was also checked to be excellent, as 10 p.p.m. of uranyl ion can be still almost completely removed, when 10 times molar excess of common competing cations are present except for the case of Ho3+, where the removal percentage was reduced to 82.8% (Fig. 7c, Supplementary Tables 7 and 8). This is due to the similar effective charge density between UO22+ and Ho3+. In addition, intensive greenish emission can be probed for the uranium-sorbed single crystals of SZ-2 when excited by 365 nm light, revealing a sharp difference from the blue emission of SZ-2 without uranium (Fig. 7d). To investigate the uranium uptake mechanism, integrated analysis of X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) on a uranium-sorbed sample of SZ-2 were measured, giving similar spectra with that obtained from aqueous solutions containing hydrated uranyl(VI) cation (Fig. 7e,f; Supplementary Table 9). Therefore, the ion-exchange process between C4mpyr+ and hydrated uranyl cations may play a major role in the sorption mechanism, which is also confirmed by X-ray photoemission spectroscopy (XPS) (Supplementary Table 10) analysis suggesting the presence of a single uranyl(VI) hydrate component in the uranyl-sorbed sample (Supplementary Fig. 23).

(a) Effect of contact times on uranyl(VI) sorption onto SZ-2 at pH 4.5 and 1.0 under stirring, with C0=10 p.p.m., mV−1=1 mg ml−1; (b) the sorption data fitted by Langmuir (green solid) and Freundlich (yellow dash) models at pH=4.5, respectively; (c) competitive sorption of coexistent ions on SZ-2 at pH=4.5, with molar ratio of metal ions to uranyl cations are ∼10 times; (d) fluorescence spectra of SZ-2 (blue) and SZ-2-U (yellow-green) excited by 365-nm light before and after uranyl adsorption; (e) XANES and EXAFS (inset) spectra of SZ-2-U, compared with the hydrated uranyl cation in aqueous solutions; (f) Fourier-transformed space (R space) spectra of SZ-2-U, compared with the hydrated uranyl cation in aqueous solution.
SZ-3 was also evaluated for uranium uptake showing a similar capability with that of SZ-2. As shown in Figure 8, the uranium removal percentage can reach 46.4% and 97.4% at pH 1.0 and 4.5, respectively. The sorption kinetics of uranyl(VI) ion with SZ-3 was also evaluated. At pH 4.5, 80.2% of uranyl ions was removed within 60 min and the sorption equilibrium with ca. 97.4% removal ratio was achieved within 3 h (Fig. 8a). At pH 1.0, the sorption equilibrium can be reached within 7 h. Sorption isotherm data fitted by the Langmuir model gives the saturated uranium sorption capacity of 58.18 mg g−1 for SZ-3 (Fig. 8b). The uranyl sorption selectivity by SZ-3 is also similar to that of SZ-2, as 10 p.p.m. of uranyl ion can be still almost completely (>95%) removed when 10 times molar excess of common competing cations are present (Fig. 8c).

(a) Effect of contact times on uranyl(VI) sorption onto SZ-3 at pH 4.5 and 1.0 under stirring, with C0=10 p.p.m., mV−1=1 mg ml−1; (b) the sorption data fitted by Langmuir (green solid) and Freundlich (yellow dash) models at pH=4.5, respectively; (c) competitive sorption of coexistent ions on SZ-3 at pH=4.5, with molar ratio of metal ions to uranyl cations are ∼10 times.
Molecular dynamics simulations on uranyl uptake
Moreover, all-atom molecular dynamics (MD) simulations were further performed to investigate the adsorption process of uranyl cation into SZ-2 and its underlying molecular mechanism. To enhance sampling, two orthogonal initial configurations were adopted to mimic the two different directions of uranyl cation adsorption: one from the top ([001] direction, Fig. 9a,b) and one from the side ([100] direction, Supplementary Fig. 26c,d). Each configuration system was simulated for six independent runs, with each 100 ns, resulting in a total aggregate simulation time of 1.2 μs. These extensive simulations reveal that the equatorial water molecules in the hydrated uranyl cation played a crucial role in mediating its binding with SZ-2 through a hydrogen bond network with those dangling hydrogen bonding acceptors from the main framework in SZ-2, such as F and O atoms that originally bind to Zr and P (Fig. 9c, Supplementary Figs 27, and 28).

The top (a) and side (b) view of the simulation system-1 (uranyl cation approaching along the c axis); (c) the final snapshot (at t=100 ns) of run 1 (out of total 6) to show the importance of equatorial water of uranyl cation in mediating its binding to the SZ-2 (the blue dash line denotes the hydrogen bond between equatorial water molecules and the dangling hydrogen bond acceptors); (d) time evolution of the electrostatic and vdW interaction energies of uranyl cation with SZ-2 and water; (e) the number of equatorial water molecules of uranyl cation (pink curve) and the number of hydrogen bonds formed between equatorial coordinating water molecules and other acceptors (including F and O in main framework) as the function of simulation time.
To further identify the driving forces for this fast sorption kinetics, we computed time evolutions of nonbonded interactions energies (including both vdW and electrostatic) between uranyl cation with SZ-2 and water (Fig. 9d). At the first 5 ns, uranyl cation was rapidly sorbed into the framework of SZ-2. During this period, the electrostatic interaction energy between uranyl cation and SZ-2 decreased from 0 to ∼−150 kJ mol−1 (Fig. 9d, black curve), and the vdW interaction energy lowered from 0 to ∼−5 kJ mol−1 (Fig. 9d, red curve), respectively. Meanwhile, the electrostatic interaction energy between uranyl cation and water increased from −1000 to∼−900 kJ mol−1, and the vdW interaction energy nearly maintained at a constant value at ∼108 kJ mol−1. This indicates that the favourable direct nonbonded interactions between uranyl cation and SZ-2 (totally gains ∼155 kJ mol−1 energy) can overcompensate for the penalty in the unfavourable interactions between uranyl cation and water (totally loses ∼100 kJ mol−1 energy), when an uranyl cation was captured by SZ-2. After entering the pores of SZ-2, few hydrogen bonds (2.38±1.03 hydrogen bonds on average, Fig. 9e blue curve) quickly formed by the equatorial water molecules in the hydrated uranyl cation with the hydrogen bonding acceptors from the main framework in SZ-2. This is thermodynamically unfavourable and a round of configuration re-adjustment (from ∼5 ns to 17 ns) quickly occurred driven by formation of a much denser hydrogen bond network through uranyl cation’s equatorial water molecules with the dangling hydrogen bonding acceptors in SZ-2. During this process, the electrostatic interaction energy between uranyl cation and SZ-2 even undergoes a remarkable increase (increased to∼−50 kJ mol−1, Fig. 9d black curve). Nevertheless, after ∼17 ns, along with the formation of more hydrogen bonds (4.15±0.79 hydrogen bonds on average, Fig. 9e blue curve) between equatorial water molecules and the dangling hydrogen bonding acceptors in SZ-2, the electrostatic interaction energy between uranyl cation and SZ-2 again decreased to ∼−150 kJ mol−1 (Fig. 9d black curve). Therefore, the strong electrostatic interaction between uranyl cation and SZ-2 effectively drives uranyl cations into the framework of SZ-2, while formation of a much denser hydrogen bond network results in a remarkably tight trapping of uranyl cation. These observations obtained from the MD simulations provided a microscopic picture of the binding with atomic details, which also support our infrared spectra analyses of SZ-2 and SZ-2-U (Supplementary Fig. 24) and the proposed ion-exchange mechanism.
Discussion
In conclusion, by utilizing a tetra-phosphonate ligand with a sterically demanding spacer and ionothermal synthesis to modify the crystallization kinetics, we obtained three highly crystalline, microporous and ultrastable zirconium phosphonate MOF compounds, the structures of which can be facilely solved by single crystal X-ray diffraction. Notably, all three compounds are built based on zero-dimensional clusters acting as the node, resembling typical SBUs found in carboxylate-based MOFs43,44, potentially making the concept of reticular chemistry available in the zirconium phosphonate system45. This is also responsible for the enhanced structural porosities observed in all three compounds compared to the previously reported zirconium phosphonate MOFs. SZ-1 represents one of the most stable MOF materials in fuming acids reported to date, and SZ-2 exhibits a superior potential for the removal of uranyl(VI) ions from aqueous solutions over a wide pH range, representing a clear improvement over the traditional inorganic zirconium phosphate ion-exchange materials, especially in the low pH region. The combined high porosity and ultrastability in fuming acids is achieved in SZ-3 with the aid of ligand distortion reduction. We believe this work may open up a new avenue to create a series of highly robust MOFs materials based on high-valence metal cations and phosphonate ligands that are currently facing a significant crystallization and designability issue but may find various potential applications under harsh conditions.
Methods
Materials and measurements
All chemicals were obtained from commercial sources (J&K Chemical, Sinopharm Chemical Reagent, Alfa Aesar, and Adamas Company) and used without further purification. 1H-NMR spectra were measured using a Unity INOVA 400 instrument. Single crystal X-ray diffraction data were collected on a Bruker D8-Venture single crystal X-ray diffractometer equipped with a Turbo X-ray Source (Mo–Kα radiation, λ=0.71073 Å) adopting the direct-drive rotating anode technique. Powder X-ray diffraction patterns were measured on a Bruker D8 advance X-ray diffractometer with Cu–Kα radiation (λ=1.54056 Å) equipped with a Lynxeye one-dimensional detector. X-ray absorption spectra were collected at the beamline 14 W of the Shanghai Synchrotron Radiation Facility. Scanning electron microscopy (SEM) images and energy-dispersive spectroscopy (EDS) data were collected on a FEI Quanta 200FEG instrument. Fluorescence spectra were obtained on a Craic Technologies microspectrophotometer. Infrared spectra were collected on a Thermo Scientific Nicolet iS50 FT-IR instrument at room temperature. The concentration of uranium was determined using a Thermo Finnigan high resolution magnetic sector Element 2 inductively coupled plasma mass spectrometer (ICP-MS). The XPS data were recorded on a PHI Quantera SXM spectrometer using monochromatic Al Kα (1486.6 eV) X-ray radiation at room temperature. The anode was operated at 24.2 W with a typical spot size of 100 μm. The U 4 f data were analysed with MultiPak software using the iterated Shirley background and the asymmetric peak profile for both primary and satellite peaks.
Colourless crystals of three compounds were mounted on Cryoloops with paratone and optically aligned by a digital camera on a Bruker D8-Venture single crystal X-ray diffractometer. The diffraction data were collected by a CMOS area detector with a Turbo X-ray Source adopting the direct-drive rotating anode technique. The data was reduced by SAINTplus package of Bruker and the structures were solved by the direct method and refined on F2 by full-matrix least-squares methods using SHELXTL (2016)46. All the non-hydrogen atoms were refined anisotropically and hydrogen atoms except those attached to water molecules, cations of ionic liquid or disordered carbons were located on calculated positions. The positional disorder is found in Tppm-ester and SZ-1 compounds, which results in relatively large values of R1 and wR2, some C level alerts in the checkcif reports, and a B level alert in compound Tppm-ester. For SZ-2, cations of ionic liquid and solvent molecules in the channels are highly disordered and impossible to refine using conventional discrete-atom models. Therefore, contributions of these molecules were removed using SQUEEZE routine of PLATON47; Structure of SZ-2 was then refined again using the data generated. For SZ-3, phenomenon of pseudo-merohedral twins was found, and was solved in monoclinic space group and contributions of disordered ionic liquid molecules were removed using SQUEEZE routine of PLATON47. CHN elemental and thermogravimetric analysis were used to determine the formula of those compounds.
Synthesis of TppmH8 and TppaH8
The synthesis route of TppmH8 and TppaH8 are shown in Supplementary Figs 1 and 2. The 1HNMR and infrared spectra for the synthesis are listed in Supplementary Figs 3, 4, 5, 6, 7, 8 and 9.
Synthesis of SZ-1
A mixture of ZrCl4 (0.0625, mmol, 0.0172, g), TppmH8 (0.025 mmol, 0.0162, g), N-butyl-N-methylpyrrolidinium bromide [C4mpyr][Br] (0.2 mmol, 0.0442, g) and 5 drops of HF (40%), was placed in a 15 ml Teflon–lined stainless steel vessel and heated at 180 °C for 3 days and then cooled to 25 °C. Colourless plate crystals were collected as a pure phase (Supplementary Figs 12 and 14). Yield: 51.4% (based on zirconium). Elemental analysis for SZ-1 (C34H36F6NO12P4Zr2.5) (Calc.: C: 36.57, H: 3.25, N: 1.25; Found: C: 37.09, H: 4.049, N: 1.516.)
Synthesis of SZ-2
SZ-2 was synthesized by the diffusion method shown in Supplementary Fig. 10: ZrCl4 and TppmH8 ligand were initially placed at the opposite sides of the reaction vessel without contact while the solid of ionic liquid [C4mpyr][Br] (2 mmol, 0.4421, g) and HF (2 drops) were located in the middle. As the temperature is evalated, ionic liquid is liquified and dissolves ZrCl4 and TppmH8 ligand, which slowly diffuse into each other and react. This substantially avoids the fast crystallization of SZ-1 products and results in the formation of SZ-2, as a pure phase with the yield of about 32.7% (based on zirconium, Supplementary Figs 13 and 15).
Synthesis of SZ-3
A mixture of ZrCl4 (0.0625, mmol, 0.0174, g), TppaH8 (0.025 mmol, 0.0193, g), N-ethylpyridinium bromide [C2py][Br] (2.1 mmol, 0.3950, g), and 2 drops of HF (40%), was placed in a 15 ml Teflon-lined stainless steel vessel and heated at 160 °C for 3 days and then cooled to 25 °C. Colourless block crystals were collected as a pure phase. Yield: 43.6% (based on zirconium). SZ-3 (C48H61F9N2O18.5P4Zr3.5) (Calc.: C: 36.58, H: 3.90, N: 1.78; Found: C: 36.34, H: 4.63, N: 1.85.)
Surface area measurements
Volumetric gas sorption data for SZ-1, SZ-2 and SZ-3 were measured at 77 K using high-purity N2 gas in a liquid nitrogen bath and the detecting pressure ranges from 0 to 760 torr. CO2 sorption measurements were also performed at 195 K in a bath of dry ice and acetone. The pretreatment of three compounds were conducted as follows: all three compounds were soaked in aqueous saturated solution of LiCl, which was refreshed in every 24 h, and this process was kept for 3 days. Then, the solution was removed and the samples were washed several times by methanol. After decanting the methanol, the samples were dried under a dynamic vacuum (<10−3 torr) at room temperature overnight and then transferred into sample tubes. Before the gas adsorption measurement, the samples were activated using the ‘outgas’ function of the surface area analyser for 4 h at 100 °C.
Stability
Hydrolytic stability measurements for all three compounds were conducted. The samples were soaked in aqueous solutions at variable pHs (pH=1–14, solution of HNO3 or NaOH), sulfuric acid (3 mol l−1), and boiling water for one day. The powder X-ray diffraction results show that SZ-1, SZ-2 and SZ-3 are stable in boiling water and solution with the pH value ranging from 1 to 11. In addition, SZ-1 is stable even in a variety of fuming acid including aqua regia, oleum, concentrated HNO3 and HCl. About 100 mg of SZ-1 was used for checking the stability in fuming acid including aqua regia, oleum, concentrated HNO3 and HCl, for 12 h at room temperature. After the treatment, fuming acid was diluted and removed, and the solid sample was washed by water and ethanol for three times, individually, and further weighted after drying in an oven at 90 °C. For each case, <5% weight loss was found, demonstrating that SZ-1 is stable in the extreme acidic environment. Furthermore, large single crystals of SZ-1 can be still isolated from on the fuming acid-soaking samples. The crystal structures can be even successfully solved using the diffraction data collected on the oleum/aqua regia-soaked SZ-1 crystals, which are identical with the original structure. The stabilities of SZ-3 were checked using the same process with that of SZ-1 and the results show that they are similar to those of SZ-1, except in the case of oleum.
Uranium sorption experiments
U(VI) sorption experiments have been performed at desired pH values. For each experiment, a total of 10 mg of SZ-1, SZ-2 and SZ-3 were added into a 50 ml container. Solution of U(VI) (10 p.p.m.) was added into the containers, with the solid–liquid ratio is 1 g l−1. The mixtures were kept on a shaking incubator for desired reaction times. The suspensions from the various reactions were filtrated and internal standard solution of indium was added for final analysis of uranium concentration using ICP-MS. U(VI) sorption experiments at desired pH values were also conducted, from 0.85 to 6.38.
X-ray absorption spectra were collected at the beamline 14 W1 of the Shanghai Synchrotron Radiation Facility with a Si(111) double crystal monochromator in transmission mode for the uranium L3-edge spectra. The electron beam energy of the storage ring was 3.5 GeV, and the maximum stored current was ∼210 mA. The uranium L3-edge EXAFS data were analysed using the standard procedures in Demeter48. Theoretical EXAFS data were calculated using FEFF 9.0 (ref. 49). Fitting procedure was performed on the k3-weighted FT-EXAFS from 2 to 10 Å−1. The amplitude reduction factor S02 was fixed at 0.9 in EXAFS fits, and the shifts in the threshold energy ΔE0 were constrained to be the same value for all fitted shells.
Quantitative information was extracted by the EXAFS fitting, as shown in Fig. 7; Supplementary Table 9. Hydrated uranium atom exhibits two oxygen atoms for achieving uranyl unit and five water molecules to give a pentagonal bipyramid coordination geometry, with U-O distances are 1.77 and 2.42 Å, respectively. Interestingly, coordination geometry of the uranium absorbed in SZ-2 is quite similar to hydrated uranyl cations, except the coordination number is 4.4 instead of 5. This is likely induced by confinement effect of the channels of the framework.
Quantum mechanics and molecular dynamics simulations
Force Parameterization of SZ-2 was obtained from quantum mechanics calculations. First-principle calculations based on the density functional theory were performed using the generalized gradient approximation (GGA) with the Becke three parameters hybrid exchange-correlation functional50,51 implemented in the Gaussian 09 program52. The smallest unrepeatable unit extracted from the experimentally obtained crystal structure was chosen as the computational model, as seen in Supplementary Fig. 26. The standard Gaussian-type basis sets 6-31G* (ref. 53) was used for C, O, F, P and H atoms. To include the scalar relativistic effects, the relativistic effective core potential ECP28MWB (ref. 54) and its corresponding (8s7p6d2f1g)/[6s5p3d2f1g] valence basis set55 were selected for Zr atoms. The atomic configuration of the computational model was kept the same as the experimentally determined structure and hence a single-point energy calculation was performed. On the basis of the ground state electron density obtained by the density functional theory, the atomic charges fit to the electrostatic potential at points selected (ESP charge) according to the CHelp scheme56 (using the Chirlian–Francl model) were then deduced. To ensure the accuracy, 10-concentric layers of points were used for each atom and >7,000 points in total were used for fitting atomic charges. These atomic charges were then used for the following classical MD simulations.
The initial configuration of SZ-2 was obtained from our X-ray crystal structure with a size of (6.616 × 6.616 × 4.749 nm3), containing 6,528 atoms (Supplementary Fig. 25). The SZ-2 force field parameters were obtained from our quantum mechanics calculations. The parameters of uranyl cation were adopted from a previous literature, in which the hydration behaviours of the equatorial plane of the uranyl cation have been elaborately studied57. System-1 and system-2 are only differing in the relative position of uranyl cation to the facets of SZ-2, to mimic the adsorption of uranyl cation into the SZ-2 from two different directions: the top (Supplementary Fig. 26a,b) and the side direction (Supplementary Fig. 26c,d). Initially, the SZ-2 were solvated into two different rectangular water boxes with the size of (6.616 × 6.616 × 10.0 nm3) and (6.616 × 12.616 × 4.749 nm3), then 384 Na+ were added to neutralize both systems. After two 1 ns NVT individual equilibriums, a uranyl cation was then introduced into two water boxes, with a distance between any heavy atoms of SZ-2 at least 1.5 nm. Meanwhile, 2 Na+ were removed to balance the charges of uranyl cation (that is, to neutralize the simulation systems again; in other words, both system-1 and system-2 contain a framework fragment of SZ-2, a uranyl cation and 382 Na+). While, system-1 containing 12401 water molecules, the system-2 containing 11,454 water molecules, respectively. This fully solvated complex was then simulated with molecular dynamics simulations, which are widely used in the studies of biomolecules58,59,60,61,62,63,64 and nanomaterials65,66,67,68,69,70.
The MD simulations were performed with the software package GROMACS (version 5.0.2)71. The VMD software72 was used to analyse and visualize the simulation results. The TIP3P water model73 was used for water molecules. During the simulation, temperature was maintained at 300 K by using v-rescale thermostat and the volume of the simulation box also remained constant (NVT)74. Periodic boundary conditions were applied in all directions. The SZ-2 was frozen throughout the simulation process. The long-range electrostatic interactions were treated with the PME method75, and the van der Waals (vdW) interactions were calculated with a cutoff distance of 1.2 nm. All solute bonds were maintained constant at their equilibrium values with the LINCS algorithm76, and water geometry was also constrained using the SETTLE algorithm77. During the production runs, a time step of 2.0 fs was used, and data were collected every 5 ps. For both the system-1 and system-2, six independent of 100 ns repeats were performed. The total aggregated simulation time was >1.2 μs.
Data availability
The X-ray crystallographic coordinates for structures reported in this study are provided as cif files in Supplementary Data 1 and have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers 1450124, 1450121, 1531836, 1450122 and 1450123. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via HYPERLINK http://www.ccdc.cam.ac.uk/data_request/cif www.ccdc.cam.ac.uk/data_request/cif.
Additional information
How to cite this article: Zheng, T. et al. Overcoming the crystallization and designability issues in the ultrastable zirconium phosphonate framework system. Nat. Commun. 8, 15369 doi: 10.1038/ncomms15369 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Deria, P. et al. Ultraporous, water stable, and breathing zirconium-based metal-organic frameworks with ftw topology. J. Am. Chem. Soc. 137, 13183–13190 (2015).
Ramaswamy, P., Wong, N. E., Gelfand, B. S. & Shimizu, G. K. H. A water stable magnesium MOF that conducts protons over 10−2 S cm−1. J. Am. Chem. Soc. 137, 7640–7643 (2015).
Jiang, H.-L. et al. An exceptionally stable, porphyrinic Zr metal-organic framework exhibiting pH-dependent fluorescence. J. Am. Chem. Soc. 135, 13934–13938 (2013).
Beyzavi, M. H. et al. A hafnium-based metal-organic framework as a nature-inspired tandem reaction catalyst. J. Am. Chem. Soc. 137, 13624–13631 (2015).
Wang, Y. et al. Umbellate distortions of the uranyl coordination environment result in a stable and porous polycatenated framework that can effectively remove cesium from aqueous solutions. J. Am. Chem. Soc. 137, 6144–6147 (2015).
Devic, T. & Serre, C. High valence 3p and transition metal based MOFs. Chem. Soc. Rev. 43, 6097–6115 (2014).
Pearson, R. G. Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533–3539 (1963).
Park, K. S. et al. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl Acad. Sci. 103, 10186–10191 (2006).
Wang, K. et al. Pyrazolate-based porphyrinic metal-organic framework with extraordinary base-resistance. J. Am. Chem. Soc. 138, 914–919 (2016).
Trickett, C. A. et al. Definitive molecular level characterization of defects in UiO-66 crystals. Angew. Chem. Int. Ed. 54, 11162–11167 (2015).
Mondloch, J. E. et al. Are Zr 6-based MOFs water stable? Linker hydrolysis versus capillary-force-driven channel collapse. Chem. Commun. 50, 8944–8946 (2014).
Guillerm, V. et al. A series of isoreticular, highly stable, porous zirconium oxide based metal–organic frameworks. Angew. Chem. Int. Ed. 51, 9267–9271 (2012).
Feng, D. et al. Zirconium-Metalloporphyrin PCN-222: mesoporous metal-organic frameworks with ultrahigh stability as biomimetic catalysts. Angew. Chem. Int. Ed. 51, 10307–10310 (2012).
Wang, B. et al. Highly stable Zr(IV)-based metal–organic frameworks for the detection and removal of antibiotics and organic explosives in water. J. Am. Chem. Soc. 138, 6204–6216 (2016).
Liu, T.-F. et al. Topology-guided design and syntheses of highly stable mesoporous porphyrinic zirconium metal–organic frameworks with high surface area. J. Am. Chem. Soc. 137, 413–419 (2015).
Bai, Y. et al. Zr-based metal-organic frameworks: design, synthesis, structure, and applications. Chem. Soc. Rev. 45, 2327–2367 (2016).
Mouchaham, G. et al. A robust infinite zirconium phenolate building unit to enhance the chemical stability of Zr MOFs. Angew. Chem. 127, 13495–13499 (2015).
Benitez, I. O. et al. Monolayers as models for supported catalysts: zirconium phosphonate films containing manganese(III) porphyrins. J. Am. Chem. Soc. 124, 4363–4370 (2002).
Wang, Z., Heising, J. M. & Clearfield, A. Sulfonated microporous organic-inorganic hybrids as strong Bronsted acids. J. Am. Chem. Soc. 125, 10375–10383 (2003).
Nixon, C. M., Le Claire, K., Odobel, F., Bujoli, B. & Talham, D. R. Palladium porphyrin containing zirconium phosphonate Langmuir-Blodgett films. Chem. Mater. 11, 965–976 (1999).
Komarneni, S. & Roy, R. Use of γ-zirconium phosphate for cesium removal from radioactive waste. Nature 299, 707–708 (1982).
Taddei, M., Donnadio, A., Costantino, F., Vivani, R. & Casciola, M. Synthesis, crystal structure, and proton conductivity of one-dimensional, two-dimensional, and three-dimensional zirconium phosphonates based on glyphosate and glyphosine. Inorg. Chem. 52, 12131–12139 (2013).
Gagnon, K. J., Perry, H. P. & Clearfield, A. Conventional and unconventional metal–organic frameworks based on phosphonate ligands: MOFs and UMOFs. Chem. Rev. 112, 1034–1054 (2011).
Gao, C., Ai, J., Tian, H.-R., Wu, D. & Sun, Z.-M. An ultrastable zirconium-phosphonate framework as bifunctional catalyst for highly active CO2 chemical transformation. Chem. Commun. 53, 1293–1296 (2017).
Cavka, J. H. et al. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 130, 13850–13851 (2008).
Morris, W. et al. Synthesis, structure, and metalation of two new highly porous zirconium metal-organic frameworks. Inorg. Chem. 51, 6443–6445 (2012).
Jiang, J. et al. Superacidity in sulfated metal–organic framework-808. J. Am. Chem. Soc. 136, 12844–12847 (2014).
Zhang, X., Zhang, X., Johnson, J. A., Chen, Y.-S. & Zhang, J. Highly porous zirconium metal–organic frameworks with β-UH3-like topology based on elongated tetrahedral linkers. J. Am. Chem. Soc. 138, 8380–8383 (2016).
Yuan, S. et al. Thermodynamically guided synthesis of mixed-linker Zr-MOFs with enhanced tunability. J. Am. Chem. Soc. 138, 6636–6642 (2016).
Mondloch, J. E. et al. Vapor-phase metalation by atomic layer deposition in a metal-organic framework. J. Am. Chem. Soc. 135, 10294–10297 (2013).
Taddei, M., Costantino, F. & Vivani, R. Synthesis and crystal structure from X-ray powder diffraction data of two zirconium diphosphonates containing piperazine groups. Inorg. Chem. 49, 9664–9670 (2010).
Costantino, F., Donnadio, A. & Casciola, M. Survey on the phase transitions and their effect on the ion-exchange and on the proton-conduction properties of a flexible and robust Zr phosphonate coordination polymer. Inorg. Chem. 51, 6992–7000 (2012).
Taddei, M. et al. The first route to highly stable crystalline microporous zirconium phosphonate metal-organic frameworks. Chem. Commun. 50, 14831–14834 (2014).
Taddei, M. et al. The use of a rigid tritopic phosphonic ligand for the synthesis of a robust honeycomb-like layered zirconium phosphonate framework. Chem. Commun. 50, 5737–5740 (2014).
Borovinskii, V. A., Lyzlova, E. V. & Ramazanov, L. M. Sorption of uranium on zirconium phosphate inorganic cation exchanger. Radiochemistry 43, 84–86 (2001).
Zhuravlev, I. et al. Uranium sorption on amorphous titanium and zirconium phosphates modified by Al3+ or Fe3+ ions. J. Radioanal. Nucl. Chem. 254, 85–89 (2002).
Garcia-Gonzalez, N., Ordonez-Regil, E., Almazan-Torres, M. G., Solis, D. & Simoni, E. Speciation of U(VI) sorbed onto ZrP2O7 in the presence of citric and oxalic acid. J. Indian Chem. Soc. 89, 305–309 (2012).
Bai, Z.-Q. et al. Introduction of amino groups into acid-resistant MOFs for enhanced U(VI) sorption. J. Mater. Chem. A 3, 525–534 (2015).
Yang, W. et al. MOF-76: from a luminescent probe to highly efficient U(VI) sorption material. Chem. Commun. 49, 10415–10417 (2013).
Carboni, M., Abney, C. W., Liu, S. & Lin, W. Highly porous and stable metal–organic frameworks for uranium extraction. Chem. Sci. 4, 2396 (2013).
Manos, M. J. & Kanatzidis, M. G. Layered metal sulfides capture uranium from seawater. J. Am. Chem. Soc. 134, 16441–16446 (2012).
Feng, M.-L. et al. Efficient removal and recovery of uranium by a layered organic–inorganic hybrid thiostannate. J. Am. Chem. Soc. 138, 12578–12585 (2016).
Deng, H. et al. Large-pore apertures in a series of metal-organic frameworks. Science 336, 1018–1023 (2012).
O'Keeffe, M., Peskov, M. A., Ramsden, S. J. & Yaghi, O. M. The reticular chemistry structure resource (RCSR) database of, and symbols for, crystal nets. Acc. Chem. Res. 41, 1782–1789 (2008).
Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705–714 (2003).
Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 71, 3–8 (2015).
Spek, A. L. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C 71, 9–18 (2015).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 12, 537–541 (2005).
Rehr, J. J., Kas, J. J., Vila, F. D., Prange, M. P. & Jorissen, K. Parameter-free calculations of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 12, 5503–5513 (2010).
Becke, A. D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C. T., Yang, W. T. & Parr, R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 37, 785–789 (1988).
Frisch, M. J. et al. Gaussian 09, Revision E.01 (Gaussian, Inc., Wallingford CT, 2013).
Hehre, W. J., Ditchfie, R. & Pople, J. A. Self-consistent molecular-orbital methods.12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 56, 2257 (1972).
Andrae, D., Häußermann, U., Dolg, M., Stoll, H. & Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 77, 123–141 (1990).
Martin, J. M. L. & Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart-Dresden-Bonn relativistic effective core potentials: the atoms Ga-Kr and In-Xe. J. Chem. Phys. 114, 3408–3420 (2001).
Chirlian, L. E. & Francl, M. M. Atomic charges derived from electrostatic potentials–a detailed study. J. Comput. Chem. 8, 894–905 (1987).
Guilbaud, P. & Wipff, G. Hydration of uranyl (UO22+) cation and its nitrate ion and 18-crown-6 adducts studied by molecular dynamics simulations. J. Phys. Chem. 97, 5685–5692 (1993).
Zhou, R. H., Huang, X. H., Margulis, C. J. & Berne, B. J. Hydrophobic collapse in multidomain protein folding. Science 305, 1605–1609 (2004).
Liu, P., Huang, X. H., Zhou, R. H. & Berne, B. J. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature 437, 159–162 (2005).
Eleftheriou, M., Germain, R. S., Royyuru, A. K. & Zhou, R. Thermal denaturing of mutant lysozyme with both the OPLSAA and the CHARMM force fields. J. Am. Chem. Soc. 128, 13388–13395 (2006).
Fitch, B. G. et al. Blue Matter: Strong Scaling of Molecular Dynamics on Blue Gene/L Springer Berlin Heidelberg (2006).
Zhou, R., Das, P. & Royyuru, A. K. Single mutation induced H3N2 hemagglutinin antibody neutralization: a free energy perturbation study. J. Phys. Chem. B 112, 15813–15820 (2008).
Das, P., King, J. A. & Zhou, R. Aggregation of gamma-crystallins associated with human cataracts via domain swapping at the C-terminal beta-strands. Proc. Natl Acad. Sci. 108, 10514–10519 (2011).
Das, P., Li, J., Royyuru, A. K. & Zhou, R. Free energy simulations reveal a double mutant avian H5N1 virus hemagglutinin with altered receptor binding specificity. J. Comput. Chem. 30, 1654–1663 (2009).
Tu, Y. S. et al. Water-mediated signal multiplication with Y-shaped carbon nanotubes. Proc. Natl Acad. Sci. 106, 18120–18124 (2009).
Li, J. Y. et al. Electrostatic gating of a nanometer water channel. Proc. Natl Acad. Sci. 104, 3687–3692 (2007).
Das, P. & Zhou, R. Urea-induced drying of carbon nanotubes suggests existence of a dry globule-like transient state during chemical denaturation of proteins. J. Phys. Chem. B 114, 5427–5430 (2010).
Guo, C., Luo, Y., Zhou, R. & Wei, G. Probing the self-assembly mechanism of diphenylalanine-based peptide nanovesicles and nanotubes. ACS Nano 6, 3907–3918 (2012).
Xiu, P. et al. Urea-induced drying of hydrophobic nanotubes: comparison of different urea models. J. Phys. Chem. B 115, 2988–2994 (2011).
Zuo, G. H., Kang, S. G., Xiu, P., Zhao, Y. L. & Zhou, R. H. Interactions between proteins and carbon-based nanoparticles: exploring the origin of nanotoxicity at the molecular level. Small 9, 1546–1556 (2013).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. Model. 14, 33–38 (1996).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 7 (2007).
Selkoe, D. J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 (2001).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Miyamoto, S. & Kollman, P. A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Acknowledgements
We thank the National Science Foundation of China (21422704, 11574224, 21471107, U1532259, 11374221, 21320102003 and 11404233), the Science Foundation of Jiangsu Province (BK20140007), ‘Young Thousand Talented Program’ in China, and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions for providing financial support for this work. T.E.A.-S. was supported as part of the Center for Actinide Science and Technology, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award Number DE-SC0016568.
Author information
Authors and Affiliations
Contributions
S.W. conceived the project. T.Z. and S.W. designed and performed synthesis and characterization. Z.Y., X.D., S.L. and R.Z. performed all-atom molecular dynamics simulations. Z.L., D.G., X.W. T.Z. and Y.G. performed the uranium sorption experiments. L.Z. and J.W. obtained and analysed the XAS data. L.C. and D.S. recorded the XPS and X-ray diffraction data. Y.W. performed the gas adsorption measurements. T.Z., S.W., J.D., Z.C. and T.E.A.-S. discussed the results. T.Z. and S.W. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures, Supplementary Tables and Supplementary References (PDF 2774 kb)
Supplementary Data 1
Cif files for SZ-1, SZ-2, SZ-3, Tppm ligand and its ester precursor. (CIF 12856 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zheng, T., Yang, Z., Gui, D. et al. Overcoming the crystallization and designability issues in the ultrastable zirconium phosphonate framework system. Nat Commun 8, 15369 (2017). https://doi.org/10.1038/ncomms15369
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms15369
This article is cited by
-
Ultra-selective uranium separation by in-situ formation of π-f conjugated 2D uranium-organic framework
Nature Communications (2024)
-
Amide-functionalized MOG for effective adsorption of U(VI) and Pb(II) from nuclear wastewater
Rare Metals (2024)
-
Reconstruction of biomimetic ionic channels within covalent organic frameworks for ultrafast and selective uranyl capture
Science China Chemistry (2024)
-
Flexible interdigitated layered framework with multiple accessible active sites for high-performance CH3I capture
Science China Chemistry (2024)
-
Synthesis and characterization of ZnO–NiO composites for efficient U(VI) scavenger
Journal of Radioanalytical and Nuclear Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
