
Abstract
Sustainable future energy scenarios require significant efficiency improvements in both electricity generation and storage. High-temperature solid oxide cells, and in particular carbon dioxide electrolysers, afford chemical storage of available electricity that can both stabilize and extend the utilization of renewables. Here we present a double doping strategy to facilitate CO2 reduction at perovskite titanate cathode surfaces, promoting adsorption/activation by making use of redox active dopants such as Mn linked to oxygen vacancies and dopants such as Ni that afford metal nanoparticle exsolution. Combined experimental characterization and first-principle calculations reveal that the adsorbed and activated CO2 adopts an intermediate chemical state between a carbon dioxide molecule and a carbonate ion. The dual doping strategy provides optimal performance with no degradation being observed after 100 h of high-temperature operation and 10 redox cycles, suggesting a reliable cathode material for CO2 electrolysis.
Similar content being viewed by others

Introduction
Solid oxide electrolysers (SOEs) have been attracting great interest due to their high efficiencies in converting low-carbon fuels from renewable electrical energy1,2. They can exploit available high-temperature heat streams from nuclear plants or exhaust industry heat to maximize electrical efficiency and both thermodynamic and kinetic advantages can be anticipated because of the high operating temperatures3,4. In SOEs, using an externally applied potential, CO2 can be electrochemically converted into CO and O2− at the cathode, while the generated O2− ions transport through the electrolyte to the anode to form O2 gas1,5,6.
Currently, nickel/yttria-stabilized zirconia (Ni-YSZ) composites are the cathode of choice for high-temperature SOEs5,6. In such a composite, the percolating networks of both Ni and YSZ provide sufficient electronic and ionic conductivity, while the Ni guarantees high electrocatalytic activity towards the reduction reaction. Long-term operation with Ni-YSZ is feasible only in CO/CO2 gas mixtures, where the presence of CO maintains a reducing atmosphere7. Under realistic conditions however, reduction–oxidation (redox) cycles of Ni will inevitably occur in the cathode, ultimately leading to electrode degradation and delamination8,9. In contrast, redox-stable ceramic cathodes would offer a promising alternative for direct high-performance CO2 electrolysis. Especially materials exhibiting n-type conduction properties are expected to demonstrate improved conductivity under the strongly reducing cathode conditions. Perovskite-type doped strontium titanates, (La,Sr)TiO3+δ (LSTO+), are such materials, due to the reducibility of Ti4+ to Ti3+, and have therefore attracted a significant amount of interest within the field of SOE and fuel cell electrodes10,11. A composite cathode based on La0.2Sr0.8TiO3.1 was shown to be well adapted to direct CO2 electrolysis12, because the titanate is partially electrochemically reduced (Ti4+→Ti3+) at potentials required for CO2 reduction and the n-type electronic conduction is accordingly enhanced, but cathode performance for CO2 electrolysis is still limited by insufficient electro-catalytic activity and the weak high-temperature chemical adsorption of reactants13.
The incorporation of catalytically active metal nanoparticles through impregnation methods has proven to be an effective approach to enhance ceramic electrode activity14. However, long-term stability of nanocatalysts at high operating temperature remains a major challenge15 due to particle agglomeration leading to performance degradation16,17. An alternative method is to incorporate the metal element as a dopant within the host lattice during the synthesis of the catalyst in air, which is then exsolved at the surface in the form of catalytically active metallic nanoparticles under reducing conditions. If the composition and conditions are carefully chosen to avoid full decomposition, anchored nanoparticles can be grown on the cathode. Any possible agglomeration of exsolved Ni nanoparticles on the substrate surface can be remedied by periodically cycling from oxidizing to reducing conditions11. We have recently demonstrated the in situ growth of metal nanoparticles directly from a perovskite backbone support through control of composition, particularly by tuning deviations from the ideal ABO3 stoichiometry18. The exsolved metal nanoparticles exhibit enhanced high-temperature stability and improved coking resistance, due to a stronger metal–oxide interface resulting from an anchoring effect with the parent perovksite. The key surface effects and defect interactions of exsolution-based perovskite materials are expected to demonstrate promising catalytic functionalities19.
High-temperature CO2 electrolysis suffers from poor adsorption and activation of the reactant, due to the linear molecules lacking polarity. This is believed to cause local starvation of CO2 in SOE cathodes1,3,7,12. Currently, preferential chemical adsorption of CO2 on solid oxide materials is based on grafting solid amines, which produces an alkaline surface. However, the desorption temperature is normally below 500 °C, which is far below typical electrolyser operating temperatures20,21. Surface oxygen vacancies created next to redox active sites on solid oxide materials may provide alternative sites for CO2 chemisorption and are expected to significantly elevate the onset temperature of chemical desorption of CO2, which would benefit cathode performance22. Combining oxygen vacancies and exsolved catalytic nanoparticles should finally produce an active interface for electrocatalytic CO2 reduction.
In this work, active nanostructures are investigated on titanate surface under differing regimes of perovskite non-stoichiometry. The exsolved metal nanoparticles coupled with tailored oxygen vacancies through Cr and Mn substitution produce a strongly interactive interface. Chemical adsorption/activation of CO2 is investigated on these titanate surfaces as well as their effectiveness as cathode materials for indirect CO2 electrolysis.
Results
Crystal structure and microstructure
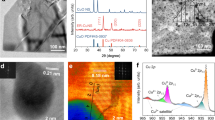
Different perovskite-defect chemistries and their combinations have been investigated, (oxygen excess, cation deficient and Cr/Mn doped) seeking to optimize electrolysis cathode properties. X-ray diffraction analysis confirms the synthesis of pure-phase La0.2Sr0.8Ti1.0O3+δ (LSTO+), La0.2Sr0.8Ti0.9Cr0.1O3+δ (LSTCO+), La0.2Sr0.8Ti0.9Mn0.1O3+δ (LSTMO+), (La0.2Sr0.8)0.95Ti0.9Mn0.1O3+δ (LSTMA−), (La0.2Sr0.8)0.95Ti0.9Cr0.1O3−δ (LSTCA−), (La0.2Sr0.8)0.95Ti0.85Cr0.1Ni0.05O3+δ (LSTCNA−) and (La0.2Sr0.8)0.95Ti0.85Mn0.1Ni0.05O3+δ (LSTMNA) powders (Fig. 1). The patterns are shown in Supplementary Section (Supplementary Fig. 1a,b). All oxidized samples could be indexed assuming a cubic symmetry, with space group Pm-3m. As shown in Supplementary Figs 1 and 2 and Supplementary Table 1, on reduction, a unit cell expansion is observed due to the reductions of Ti, Mn and Cr (from M4+ to M3+/2+). The doping and reduction of Mn/Cr should be coupled with the creation of additional oxygen vacancies in the titanate, which in turn should facilitate CO2 chemisorption. The LSTO+ is oxygen-excess while the oxygen with δ amount is present in the form of oxygen interstitial in perovskite oxide. However, the (La,Sr)(Ti,M)O3+δ (M=Mn, Cr) has redox-active dopants such as Mn linked to oxygen vacancies even though the interstitial oxygen might be still present after reduction. The nickel containing compounds LSTMNA− and LSTCNA− exhibit nickel exsolution on reduction, as observed in X-ray diffraction, X-ray photoelectron spectroscopy (XPS) and microscopy. Supplementary Fig. 2 shows the emergence of a peak at 2θ=44.5°, corresponding to cubic metallic nickel. Due to the A-site deficiency, the perovskite structure is retained for the titanate phase, suggesting structural redox stability. XPS on oxidized and reduced samples shows that nickel is only present as Ni2+ in the former, whereas metallic Ni0 is additionally present in the latter, corroborating the X-ray diffraction results. XPS furthermore confirms the presence of the reduced species Ti3+, Mn3+ and Cr3+/2+ within the reduced titanates (Supplementary Figs 3 and 4). Figure 2a,b present scanning electron microscopic micrographs of sintered LSTMNA− pellets after reduction, showing uniform titanate surface decoration with Ni nanoparticles. The nanoparticles exist within a narrow size distribution, with an average of 60 nm, which should prove highly active as electrocatalyst. The growth of these Ni nanoparticles was studied using high-resolution transmission electron microscopy. From the lattice spacings in Fig. 2c,d, 0.275 and 0.203 nm, it becomes clear that two distinct phases have emerged, that is, reduced titanate (110) and metallic nickel (111), respectively. A clear hetero-junction is visible between the two phases, suggesting a strong interaction between the titanate backbone and exsolved nanoparticles. This anchoring effect is expected to provide enhanced thermal stability of the catalyst, preventing the severe long-term agglomeration which is commonly observed for impregnated catalysts23. Regeneration of the nanoparticles is also possible through periodic redox cycling. Besides, selected-area electron diffraction analysis for the reduced LSTMNA− is shown in Supplementary Fig. 5, it shows that LSTMNA− is able to produce the polycrystalline structure.

X-ray diffraction of (a) oxidized and (b) reduced LSTO+, LSTMO+, LSTCO+, LSTMNA− and LSTCNA−; X-ray diffraction Rietveld refinement of reduced (c) LSTMNA− and (d) LSTCNA−. bckgr, background; calc, calculated; diff, difference; obs, observed.

(a) Scanning electron microscopic results for the reduced LSTMNA−, Scale bars, 5 μm; (b) Ni nanoparticles size distribution of the reduced LSTMNA− inset, Scale bars, 3 μm; (c) High-resolution transmission electron microscopic results for the reduced LSTMNA−, Scale bars, 5 nm; (d) the legible hetero-junction of Ni and substrate along the corresponding to pink square shown in (c), Scale bars, 2 nm.
Oxygen nonstoichiometry and conductivity
Thermogravimetric analysis on the various titanates, as shown in Supplementary Fig. 6 and Supplementary Table 2, suggests varying degrees of oxygen stoichiometry, depending on titanium substitution with Cr/Mn/Ni. Whereas LSTO+ shows a change of 0.032 mol of oxygen upon oxidation/reduction, the Cr- and Mn-substituted stoichiometries release/absorb 0.039 and 0.056 mol of lattice oxygen, confirming the reducibility of the Mn and consequent formation of additional oxygen vacancies upon reduction. However, the Cr3+ in titanate lattice is less reducible that leads to similar oxygen absorption for reduced LSTO+ and LSTCO+ samples. And strong reduction only produces partial transition of Cr3+ to Cr2+ as confirmed by the larger weight gains in thermogravimetric analysis (TGA) in Supplementary Fig. 6c. The weight changes increase even further upon nickel introduction and it is suggested that lattice nickel was partially reduced to metallic nickel during reduction at 800 °C for 20 h. This would result in reduced titanate with Ni present as secondary phase. Assuming that the majority of metallic Ni resides on the titanate surface, it is clear that an active nanostructure is formed with the presence of Ni nanoparticles coupled with a large concentration of oxygen vacancies.
The varying oxygen contents and abilities to store and release lattice oxygen also manifest themselves in the oxide ionic conductivities of the titanates. The ion conductivity of sintered pellets is tested using the electron-blocking electrode method for the reduced states24. As shown in Supplementary Fig. 7, reduced LSTO+ has the lowest ionic conductivity, reaching 6.8 × 10−4 S cm−1 at 800 °C. Upon substitution with Cr/Mn and consequently increasing oxygen vacancy concentration under reducing conditions, this increases by an order of magnitude to 6.1–6.3 × 10−3 S cm−1. Introducing A-site deficiency and nickel on the B-site causes an additional 30% increase to 8.6–9.3 × 10−3 S cm−1 under identical conditions. These experimental observations are further substantiated by theoretical calculations on oxide ion migration in simplified SrTiO3. These reveal that oxide ion transport by a vacancy-mediated mechanism is much more favourable than through an interstitial one; as shown in Supplementary Figs 7 and 8, the energy barrier for the former is 0.42 eV, as compared to 5 eV for the latter. This is to be expected for the dense perovskite structure and confirms that creating additional oxygen ion vacancies should indeed be beneficial for ionic transport by increasing the concentration of charge carriers.
CO2 adsorption and activation
The chemical adsorption of CO2 molecules on reduced titanate samples was investigated by in situ infrared spectroscopy. The infrared scans for all the samples without CO2 adsorption at room temperature are shown in Supplementary Fig. 9a,b, in which no CO2 or carbonate species (CO32−) signals are observed. In contrast, all samples with adsorbed CO2 show absorption signals in two distinct infrared bands at room temperature in Supplementary Fig. 9c,d, that is, 2,380–2,300 cm−1, which is associated with CO2 molecules25 on the sample surface, and 1,500–1,430 cm−1, which is typically observed for CO32− (ref. 26). The chemisorbed carbon dioxide on the titanate surface is therefore assumed to be intermediate between molecular CO2 and carbonate ions. As expected from the concentration of oxygen ion vacancies, which should translate into an active oxide surface, the strongest infrared absorption is observed for LSTMNA− and LSTCNA−. To test the ability to adsorb CO2 at elevated temperature, LSTMNA− was subjected to in situ infrared tests between 400 and 1,200 °C. As Fig. 3a,b reveal, both infrared bands remain visible up to 1,200 °C, suggesting that both CO2 and carbonate species remain adsorbed, due to strong bonding with this titanate’s surface. Temperature Programmed Desorption sheds further light on the adsorption/desorption behaviour. Figure 3c shows how physisorbed CO2 is desorbed below 100 °C, whereas chemisorbed CO2 is retained until much higher temperatures. For Cr/Mn-substituted titanate, the strongest desorption is extended to approximately 800 °C, with concomitant increased adsorption capacity by an order of magnitude as compared to LSTO+ (0.043 versus 0.0056, ml m−2, respectively). These results are further corroborated by TGA in pure Ar atmosphere on reduced titanate samples that have subsequently been treated in CO2 at room temperature for 60 min, shown in Supplementary Fig. 9e. Much enhanced weight loss and continued desorption between 800 and 1,200 °C is observed for all Cr/Mn-substituted samples as compared to LSTO+, which desorbs almost all CO2 below 800 °C. This is obviously also much improved when compared to amine-grafted oxides, which typically desorb below 500 °C (refs 20, 21).

(a,b) In situ infrared spectroscopy of CO2 adsorbed on the reduced LSTMNA− from 400 to 1,200 °C; (c) Temperature Programmed Desorption (TPD) test of CO2 on the reduced samples from 50 to 1,000 °C in pure CO2.
Theoretical calculations were carried out to construct a tentative mechanism for CO2 adsorption and activation on the titanate surface in conjunction with exsolved Ni nanaoparticles and oxide vacancy defects. Before considering the presence and effect of such oxide vacancies however, we first study the CO2 chemisorption behaviour on the boundary of a Ni cluster and SrTiO3 pristine surface (110), see Supplementary Fig. 10. As shown in Fig. 4a and Supplementary Fig. 11c, upon adsorption, low energy scenarios are found for CO2 forming a bidentate configuration including a carbonate formation (Supplementary Fig. 11a). Lowest adsorption energies of −2.23 and −2.60 eV are calculated for the configurations in which the carbon atom of CO2 binds with a Ni atom from the cluster, whereas one of CO2’s oxygen atoms (O1) attaches to an hcp site on the titanate surface, interacting with two Sr atoms and one Ti atom. With a calculated bond length of 1.906 Å for C–Ni, a strong interaction is expected. The Sr–O1 and Ti–O1 distances are 2.490 Å and 2.194 Å, respectively, which differ substantially from bulk strontium titanate (2.767 Å for Sr–O (ref. 27) and 1.952 Å for Ti–O from experimental observations28). The C–O1 bond length is elongated to 1.403 Å and the O–C–O angle is bent to 120.9° relative to the gaseous values of 1.18 Å and 180.0°, respectively. This suggests that apart from a strong interaction between adsorbed CO2 and the Ni/SrTiO3 surface, the distorted CO2 seems highly activated. Furthermore, by binding another CO2 molecule to the previously adsorbed oxygen atoms of CO2, such as O2 atom in Fig. 4a, these bidentate configurations could be basic configurations to further generate carbonate, which may play a role as shown in our infrared experimental results. Contour plots shown in Fig. 4a indicate that the Ni cluster donates electron charge to the adsorbed CO2 molecule; the charge density changes take place predominantly within the 2p orbitals of C, O, Ni and relevant surface atoms. The electrostatic interactions between surface and CO2, which would also affect the observed bidentate configuration, clearly suggest a strong chemical adsorption and activation, promoted by Ni exsolution.

Different adsorption configurations of CO2 on (a) the (110) Ni/STO system surface and (b) defected site of the (110) Ni/STO system surface. The upper panels show side views while the middle panels give top–down views. Unit cells used in the calculations are marked by blue rectangles. Nickel is blue, strontium is green, titanium is pale, oxygen is red and carbon is grey. The bottom panels show contour plots of the electronic charge density difference for CO2 adsorption on the (110) Ni/STO system surface (bottom left) and defected site of the (110) Ni/STO system surface (bottom right).
Next we consider adsorption onto a titanate surface that includes oxygen vacancy defects. We use a simplified scenario of a single oxide vacancy site residing in close proximity to the Ni cluster on the SrTiO3 (110) surface and simulate how CO2 would approach and adsorb onto this site as shown in Supplementary Fig. 12. A distinctly different adsorption mechanism emerges now as compared to the vacancy-free titanate surface: in this scenario the CO2 molecule exothermically dissociates into a CO moiety binding to the Ni cluster and an oxygen atom occupying the vacant surface site, with greatly enlarging of C–O1 distance from gaseous value of 1.177 to 3.265 Å (Fig. 4b and Supplementary Fig. 12). The carbon atom of CO2 is now binding to two Ni atoms, with bond lengths C–Ni1 of 1.820 and C–Ni2 of 1.889 Å. The calculated Sr–O1 and Ti–O1 bond distances are 2.728–2.783 Å and 2.020–1.880 Å, respectively, which are very similar to those observed in bulk SrTiO3 (2.767 and 1.952 Å, respectively27,28, indeed suggesting that O1 is now occupying the oxygen vacancy. The contour plot for this scenario shows that charge density is increased on 2p orbital of C and O1 atom and depleted on Ni 4s orbitals. These charge rearrangements mainly result from the C–O1 bond breaking that causes charge transfer back to the C and O1 atom, respectively. Moreover, electron charge density is also donated by the Ni cluster to the C 2p orbital, albeit originating from Ni 4s orbitals instead. The 2p orbital of O1 accepts charge density from the neighbouring surface atoms. These calculations strongly suggest a synergistic effect from the exsolved Ni cluster and neighbouring oxygen vacancy, causing promotion of the adsorbed CO2 molecule by effectively weakening the C–O1 bond. This promotion effect is expected to translate itself into a very catalytically active surface for CO2 electrolysis.
Electrolysis performance
Both symmetrical and full electrolyser cells were fabricated, comprising composite titanate cathodes with Ce0.8Sm0.2O2−δ. As can be seen in Supplementary Fig. 13, the cells contain dense electrolytes as well as porous electrodes of around 10 μm in thickness. AC impedance was performed on the symmetrical cells to assess the electrode polarization behaviour under reducing conditions, with pH2 ranging from 0.20 to 1.0 bar. For all titanates, it was found that the series resistance Rs was dominated by the YSZ electrolyte, confirming sufficient electronic conductivity as provided by the reduced titanates. And the ohmic resistance of electrode is negligible because the conductivity of electrodes is 3–4 orders of magnitude higher than that of the YSZ electrolyte. The electrode performance of all materials improved significantly with increasing pH2 from 0.20 to 1.0 bar, with polarization resistances decreasing by 50–60%. LSTO+ was found to have the poorest performance (5.32–2.00 Ω cm2), whereas Cr/Mn-substituted and Ni-exsolving LSTMNA− and LSTCNA− cathodes perform best, with the lowest Rp recorded of 0.67 Ω cm2 at 800 °C and pH2=1.0 bar. The symmetrical cell results are summarized in Supplementary Figs 14–16.
The cells were used to perform direct CO2 electrolysis under varying applied voltages, ranging from 1.2 to 2.0 V at 800 °C. The open circuit voltage of the cells was established by exposing the cathode and anode to 100% H2 and static air, respectively, and was found to reach 1.1 V, indicating good gas separation by the cells. Typical current–voltage (I–V) curves for the various cathode materials in direct CO2 electrolysis mode can be found in Fig. 5. As expected from the previous findings, LSTO+ shows the poorest performance, reaching a maximum current density of 0.24 A cm−2 at 2.0 V. Improved performance is observed for Mn/Cr-substituted compositions, which can be attributed to enhanced CO2 adsorption and activation at high temperatures. LSTMO+ reaches a maximum current density of 0.53 A cm−2 at 2.0 V. The nickel exsolving compositions show a further improvement, with maximum current densities of 0.91 and 0.79 A cm−2 at 2.0 V for LSTMNA− and LSTCNA−, respectively. As the potentiostatic measurement in Fig. 5b shows, performances tail off somewhat over time, but a stable current density of 0.87 A cm−2 seems to be attained for LSTMNA− at 2.0 V, a factor 3–4 times higher than observed for LSTO+. Further evidence of these titanates’ suitability to reduce CO2 at the cathode comes from inspecting the rate of CO production as measured by gas chromatography and corresponding Faradaic efficiencies at different applied voltages, shown in Fig. 5c,d. LSTMNA− and LSTCNA− yield 6.08 and 5.16 ml min−1 cm−2 of CO at 2.0 V and 800 °C, respectively, corresponding to Faradaic efficiencies of 99.9 and 98.4%, respectively. When compared to unmodified LSTO+, this constitutes a 400% improvement of performance. The effect of Ni exsolution yields a performance boost of 75–85% when comparing with Cr/Mn substitution only. Results have been summarized in Table 1.

(a) I–V curves of CO2 electrolysis with different cathodes at 800 °C; (b) short-term performances of CO2 electrolysis at different voltages; (c) CO production and (d) current efficiency with different cathodes.
One aspect that has been ignored so far is the effect of A-site deficiency on electrode performance. To enhance Ni exsolution, 5% A-site deficiency was introduced in the LSTMNA− and LSTCNA− materials, but the A-site deficiency itself may enhance electrode performance. The oxygen interstitial is present in LSTO+, LSTMO+ and LSTCO+, though the oxygen vacancies are created and linked to redox active dopants such as Mn/Cr after reduction. However, the A-site deficiency would further produce higher oxygen vacancy concentration that may be positive to electrode activity enhancement. To study this effect, Cr/Mn-substituted LSTMA− and LSTCA− (5% A-site deficiency but no nickel substitution) were also used as cathode materials. Their performance, as indicated in Supplementary Fig. 17, is in between LSTMO+/LSTCO+ and LSTMNA−/LSTCNA−, suggesting a positive electrocatalytic effect from the A-site deficiency. This may be the result of enhanced reducibility in such compositions and hence oxygen vacancy concentration, as proposed by Savaniu and Irvine29. Still, the effect of Ni exsolution adds to the cathode’s electrochemical activity, as indicated by the superior performance.
To further understand the observed electrochemical performance in CO2 electrolysis mode at 800 °C for the different cathode materials, impedance spectroscopy was carried out under a range of applied biases, that is, 1.1–1.6 V. Similar trends are observed for all cathode materials, as shown in Supplementary Figs 18 and 19. First, series resistance, Rs, is stable and dominated by the electrolyte resistance, which is indicative of the titanates’ capability to provide an electronically conductive electrode backbone. In our full electrolysers, the ohmic resistance of electrode is negligible because the conductivity of titanates is 3–4 orders of magnitude higher than that of YSZ electrolyte. Second, polarization resistances, Rp, decrease with increasing voltages, which may be due to that the increasing potential leads to a stronger reducing potential that produces higher oxygen vacancy concentration in titanate and thus better ionic conductivity and CO2 adsorption/activation, enhancing electrode kinetics. A change in the fermi level of the electron can also be expected to facilitate electrode polarizations by increasing applied potential. The electrode response seems to be dominated by two low frequency processes. One is related to gas conversion and its response diminishes upon increasing the applied voltage, as expected. The second process is tentatively attributed to dissociative adsorption of reactant species. This seems justified, as Rp is largest for LSTO+ (4.92–1.73 Ω cm2 at 1.1–1.6 V) and decreases for LSTMO+ (2.86–0.82 Ω cm2 at 1.1–1.6 V) to finally be lowest for LSTMNA− (1.41–0.51 Ω cm2 at 1.1–1.6 V). Therefore Rp seems to decrease with the electrode’s increasing ability to chemically adsorb and activate CO2, with the best electrode performance observed for electrodes with both exsolved Ni and oxygen vacancies on its surface. The hetero-junction interface with Ni nanoparticles interacting with oxygen vacancy on titanate provides a synergy to facilitate electrode reaction. However, the catalytic activity of Mn itself in ABO3 oxide is better than the Cr-doped sample, and the ionic conductivity of LSTMO+ is higher than that of LSTCO+. The above two points would accordingly deliver better performance for LSTMO+ electrode even though LSTCO+ has a higher oxygen vacancy concentration. The different cathode materials’ impedance responses at 1.6 V are summarized in Fig. 6.

(a) The comparison of Rp for the high-temperature CO2 electrolysis with different electrode; (b) in situ AC impedance at 1.6 V.
The long-term stability of the exsolving cathode material LSTMNA− was assessed by performing CO2 electrolysis for a period of 100 h at 1.3 V and 800 °C in 100% CO2 atmosphere. As shown in Fig. 7a, a stable current density of 0.28 A cm−2 is retained for the duration of the experiment. The stability of this material upon switching between oxidizing and reducing conditions was similarly assessed by redox cycling at 800 °C in alternating 5% H2/Ar and air atmospheres, with subsequent CO2 electrolysis at 1.3 V. As indicated by Fig. 7b and Supplementary Fig. 20, no performance degradation can be observed after 10 such redox cycles, demonstrating excellent stability of the active nanostructured surface of the titanate within the frames of these experiments. The anchoring of Ni nanoparticles to the perovskite lattice is expected to improve both long-term and redox stability, by preventing particle coalescence driven by surface energy reduction18.

(a) The long-term performance and current efficiency of the LSTMNA− cathode with the flow of CO2 at 800 °C; (b) the short-term performance of LSTMNA− cathode after 10 redox cycles.
Discussion
In conclusion, we have shown exceptionally high performance for direct CO2 electrolysis with near 100% Faradaic efficiency. The key is the tailored surface structure of doped strontium titanate that is in situ constructed through a combination of Ni particle exsolution and oxygen stoichiometry engineering. These active surface structures enable high-temperature chemical adsorption/activation of CO2 and furthermore exhibit high-temperature stability for several tens of hours with significant redox cycling ability. Our work shows that these titanates are serious candidates for alternative cathode materials in SOE cells, providing the tools to develop the next generations of electrochemical devices for energy conversion and storage.
Methods
Synthesis
Ceramic oxides including La0.2Sr0.8Ti1.0O3+δ(LSTO+), La0.2Sr0.8Ti0.9Cr0.1O3+δ (LSTCO+), La0.2Sr0.8Ti0.9Mn0.1O3+δ(LSTMO+), (La0.2Sr0.8)0.95Ti0.9Mn0.1O3+δ (LSTMA−), (La0.2Sr0.8)0.95Ti0.9Cr0.1O3−δ (LSTCA−), (La0.2Sr0.8)0.95Ti0.85Cr0.1Ni0.05O3+δ (LSTCNA−), (La0.2Sr0.8)0.95Ti0.85Mn0.1Ni0.05O3+δ (LSTMNA−), (La0.8Sr0.2)0.95MnO3−δ (LSMA−) and Ce0.8Sm0.2O2−δ powders were synthesized using a solid-state reaction method performed in air30.
Characterization
Phase formations were confirmed by using X-ray diffraction (Cu Kα, Miniflex 600, Rigaku Corporation, Japan), and the data were refined by using the General Structure Analysis System software31. Scanning electron microscopy (SU-8010, JEOL Ltd, Japan) and high-resolution transmission electron microscopy (Tecnai F20, FEI Ltd, USA) were employed to investigate the exsolution of nanoparticles. XPS (ESCALAB 250Xi, Thermo, USA) with monochromatized Al Kα at hv=1,486.6 eV was utilized to analyse elemental oxidation states. TGA was conducted on a Netzsch STA449F3 to calculate oxygen non-stoichiometry. To this end, powder samples were first reduced in 5% H2/Ar at 800 °C for 20 h and TGA was subsequently performed on these samples from room temperature to 1,200 °C in air. The electrical properties of the reduced samples were examined using DC four-terminal method (Keithley 2,000, Keithley Instruments Inc., USA) in an atmosphere of 5% H2/Ar between 650 and 800 °C. The H2O content in 5%H2/Ar or H2 streams were measured to be 0.5 and 1%, respectively corresponding to oxygen partial pressures of ∼1 × 10−20 and ∼1 × 10−22 atm, respectively, at 800 °C. Approximately 2.0 g of the samples’ powders was pressed into pellets and sintered at 1,400 °C for 10 h in air to get dense samples for the conductivity tests. Before the conductivity tests, the samples’ pellets were reduced at 800 °C for 20 h in 5% H2/Ar. All infrared spectra were collected using Fourier transform infrared spectrometer (VERTEX 70, Bruker). The powder samples were first reduced in 5% H2/Ar at 800 °C for 20 h and Temperature Programmed Desorption tests of CO2 were subsequently recorded from 50 to 1,200 °C in CO2 with a Micromeritics-Hiden Autochem II 2920-QIC20.
Electrochemical characterization
Symmetric cells with 0.5-mm-thick YSZ and different titanate electrodes (1 cm2) were assembled and treated at 1,200 °C for 3 h in air. The current collector was made with silver paste (SS-8060, Xinluyi, China) and treated at 550 °C for 30 min in air. The 0.5-mm-thick YSZ-supported single SOEs with titanate cathodes (1 cm2) and LSM anode (1 cm2) were assembled and treated at 1,200 °C for 3 h in air. The current collector was made with silver paste (SS-8060, Xinluyi, China) and treated at 550 °C for 30 min in air. Electrochemical measurement was performed using an electrochemical station (IM6, Zahner, Germany). The frequency range was 4 MHz to 100 mHz, and the voltage perturbation was 10 mV. The gas flow was controlled with mass flow meters (D08-3 F, Sevenstar, China).The electrolysis experiment was conducted by flowing 5% H2/Ar at the flow rate of 50 ml min−1 for 1.5 h and then pure hydrogen at the flow rate of 50 ml min−1 was supplied to cathode for 2.0 h at operation temperature. After that, the cathode would be sufficiently reduced and activated. And then the CO2 electrolysis was performed. The online gas chromatography (GC2014, Shimazu, Japan) was used to analyse the CO production of the output gas from the electrolyser cells at the flow rate of 50 ml min−1 at 800 °C.
Theoretical calculations
Theoretical calculations are utilized to understand the mechanism of oxide ion conduction and CO2 chemical adsorption. All calculations were performed using density functional theory implemented in the Vienna Ab Initio Simulation Package (VASP)32,33. Within the projector augmented wave (PAW) framework, the plane-wave cutoff energy was set to 500 eV, which gives well-converged relative energies for the system. The Perdew–Burke–Ernzerhof functional was used to describe exchange and correlation34. In electronic and geometric optimizations, energies and residual forces were converged to 10−6 eV and 0.02 eV Å−1, respectively. In order to simplify and facilitate the calculation, the creation of oxygen vacancies through La and Mn/Cr substitution on the A and B-site, respectively, has been simplified by making oxygen-deficient SrTiO3. The lattice parameters of SrTiO3 (STO), optimized with a 13 × 13 × 13 k-point grid35, were a=b=c=3.947 Å, which is in good agreement with our experimental values. A superstructure with dimensions of 11.86 × 11.86 × 7.89 Å3 (89 atoms) was used to simulate the oxygen transport process within STO bulk. A 2 × 2 × 4 k-point grid was used for Brillouin zone sampling of this superstructure model. The climbing-image nudged elastic band method36 was used to explore the transition states of the oxygen vacancies transport process. The periodic slab model was used to simulate the (110) surface of STO with Sr-Ti-O terminations. For this model, three Sr-Ti-O layers and two O layers were included in the slab. The three bottom layers were fixed to their bulk geometries during optimization while other atoms were fully relaxed. A vacuum layer with thickness of about 22 Å was inserted in the c direction to avoid the slab-to-slab interaction. The Ni segregation on the (110) surface slab of STO was mimicked by a system containing a Ni cluster with 11 Ni atoms laying on (110) surface of STO with Sr-Ti-O terminations. A 2 × 2 × 1 k-point grid is employed for Brillouin zone sampling of (110) Ni/STO surface system. The adsorption energy of CO2 is calculated as E=Etot−Eslab−Ead, where Etot, Eslab and Ead are the energy of the Ni/STO surface system without adsorption, the energy of the CO2 in gas phase and the total energy of the adsorption system, respectively.
Data availability
The data that support the findings of this study are available from the corresponding author(s) upon request.
Additional information
How to cite this article: Ye, L. et al. Enhancing CO2 electrolysis through synergistic control of non-stoichiometry and doping to tune cathode surface structures. Nat. Commun. 8, 14785 doi: 10.1038/ncomms14785 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Yue, X. & Irvine, J. T. S. Alternative cathode material for CO2 reduction by high temperature solid oxide electrolysis cells. J. Electrochem. Soc. 159, F442–F448 (2012).
LagunaBercero, M. A., Skinner, S. J. & Kilner, J. A. Performance and characterization of (La, Sr)MnO3/YSZ and La0.6Sr0.4Co0.2Fe0.8O3 electrodes for solid oxide electrolysis cells. Chem. Mater. 22, 1134–1141 (2010).
Xie, K. et al. Direct synthesis of methane from CO2/H2O in an oxygen-ion conducting solid oxide electrolyser. Energy Environ. Sci. 4, 2218–2222 (2011).
Hauch, A. et al. Highly efficient high temperature electrolysis. J. Mater. Chem. 18, 2331–2340 (2008).
Zhan, Z. & Lin, Z. Electrochemical reduction of CO2 in solid oxide electrolysis cells. J. Power Sources 195, 7250–7254 (2010).
Knibbe, R. et al. Solid oxide electrolysis cells: degradation at high current densities. J. Electrochem. Soc. 157, B1209–B1217 (2010).
Ebbesen, S. D. & Mogensen, M. Electrolysis of carbon dioxide in solid oxide electrolysis cells. J. Power Sources 193, 349–358 (2009).
Bidrawn, F. et al. Efficient reduction of CO2 in a solid oxide electrolyzer. Electrochem. Solid-State Lett. 11, 167–170 (2008).
Kim-Lohsoontorn, P. & Bae, J. Electrochemical performance of solid oxide electrolysis cell electrodes under high-temperature coelectrolysis of steam and carbon dioxide. J. Power Sources 196, 7161–7168 (2011).
Tsekouras, G. & Irvine, J. T. S. The role of defect chemistry in strontium titanates utilised for high temperature steam electrolysis. J. Mater. Chem. 21, 9367–9376 (2011).
Tsekouras, G., Neagu, D. & Irvine, J. T. S. Step-change in high temperature steam electrolysis performance of perovskite oxide cathodes with exsolution of B-site dopants. Energy Environ. Sci. 6, 256–266 (2012).
Li, Y. et al. Composite fuelelectrode La0.2Sr0.8TiO3−δ–Ce0.8Sm0.2O2−δ for electrolysis of CO2 in an oxygen-ion conducting solid oxide electrolyser. Phys. Chem. Chem. Phys. 14, 15547–15553 (2012).
Yang, L. et al. A composite cathode based on scandium doped titanate with enhancedelectrocatalytic activity towards direct carbon dioxide electrolysis. Phys. Chem. Chem. Phys. 16, 21417–21428 (2014).
Xing, R. et al. Co-electrolysis of steam and CO2 in a solid oxide electrolysis cell with La0.75Sr0.25Cr0.5Mn0.5O3−δ –Cu ceramic composite electrode. J. Power Sources 274, 260–264 (2015).
Kim, G. et al. Investigation of the structural and catalytic requirements for high-performance SOFC anodes formed by infiltration of LSCM. Electrochem. Solid-State Lett. 12, B48–B52 (2009).
Xu, S. et al. Composite cathode based on Fe-loaded LSCM for steam electrolysis in an oxide-ion-conducting solid oxide electrolyser. J. Power Sources 239, 332–340 (2013).
Li, Y. et al. Composite cathode based on Ni-loaded La0.75Sr0.25Cr0.5Mn0.5O3−δ for direct steam electrolysis in an oxide-ion-conducting solid oxide electrolyzer. Int. J. Hydrogen Energy 38, 10196–10207 (2013).
Neagu, D. et al. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 5, 916–923 (2013).
Neagu, D. et al. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 6, 8120 (2015).
Hicks, J. C. et al. Designing adsorbents for CO2 capture from flue gas-hyperbranched aminosilicas capable of capturing CO2 reversibly. J. Am. Chem. Soc. 130, 2902–2903 (2008).
Khatri, R. A. et al. Thermal and chemical stability of regenerable solid amine sorbent for CO2 capture. Energy Fuels 20, 1514–1520 (2006).
Qi, W. et al. Remarkable chemical adsorption of manganese-doped titanate for direct carbon dioxide electrolysis. J. Mater. Chem. A 2, 6904–6915 (2014).
Irvine, J. T. S. et al. Evolution of the electrochemical interface in high-temperature fuel cells and electrolysers. Nature Energy 1, 15014 (2016).
Li, X. et al. Synthesis and electrical properties of Co-doped Y0.08Sr0.92TiO3−δ as a potential SOFC anode. Solid State Ionics 179, 1588–1592 (2008).
Figueiredo, M. C. et al. In situ spectroscopic study of CO2 electroreduction at copper electrodes in acetonitrile. ACS Catal. 6, 2382–2392 (2016).
Su, W. et al. Surface phases of TiO2 nanoparticles studied by UV Raman spectroscopy and FT-IR spectroscopy. J. Phys. Chem. C 112, 7710–7716 (2008).
Kuroiwa, Y. et al. Atomic replacement effect on electron charge density of perovskite oxides. J. Korean Phys. Soc. 42, 1425–1428 (2003).
Silva, L. F. et al. Long-range and short-range structures of cube-like shape SrTiO3 powders: microwave-assisted hydrothermal synthesis and photocatalytic activity. Phys. Chem. Chem. Phys. 15, 12386–12393 (2013).
Savaniu, C. D. & Irvine, J. T. S. Reduction studies and evaluation of surface modified A-site deficient La-doped SrTiO3 as anode material for IT-SOFCs. J. Mater. Chem. 19, 8119–8128 (2009).
Gan, Y. et al. Composite cathode La0.4Sr0.4TiO3−δ–Ce0.8Sm0.2O2−δ impregnated with Ni for high-temperature steam electrolysis. J. Power Sources 245, 245–255 (2014).
Tao, S. & Irvine, J. T. S. Structural and electrochemical properties of the perovskite oxide Pr0.7Sr0.3 Cr0.9Ni0.1O3−δ . Solid State Ionics 179, 725–731 (2008).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. ERRATA: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
K.X. acknowledges Natural Science Foundation of China (91545123) and Natural Science Foundation of Fujian Province (2016J01275) for funding this work. C.L. acknowledges support by the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB20000000 and Hundred Talents Program of the Chinese Academy of Sciences. J.T.S.I. acknowledges funding from EPSRC Platform Grant EP/K015540/1 and Royal Society Wolfson Merit Award WRMA 2012/R2.
Author information
Authors and Affiliations
Contributions
K.X. fully designed and supervised the project. J.T.S.I. supervised part of the project. L.Y. conducted the experimental work. M.Z. and C.L. carried out the theoretical calculations. All authors contributed to data analysis and gave approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures and Supplementary Tables (PDF 3470 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ye, L., Zhang, M., Huang, P. et al. Enhancing CO2 electrolysis through synergistic control of non-stoichiometry and doping to tune cathode surface structures. Nat Commun 8, 14785 (2017). https://doi.org/10.1038/ncomms14785
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms14785
This article is cited by
-
Nanoparticle Exsolution on Perovskite Oxides: Insights into Mechanism, Characteristics and Novel Strategies
Nano-Micro Letters (2024)
-
Self-assembled FeRu bimetallic nanocatalysts for efficient and durable mutual CO–CO2 conversion in a reversible solid oxide electrochemical cell
Science China Materials (2024)
-
Real-time insight into the multistage mechanism of nanoparticle exsolution from a perovskite host surface
Nature Communications (2023)
-
Tandem engineering for CO2 electrolysis toward multicarbon products
Nano Research (2023)
-
Boosting the stability of perovskites with exsolved nanoparticles by B-site supplement mechanism
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
