
Abstract
3D porous nanostructures built from 2D δ-MnO2 nanosheets are an environmentally friendly and industrially scalable class of supercapacitor electrode material. While both the electrochemistry and defects of this material have been studied, the role of defects in improving the energy storage density of these materials has not been addressed. In this work, δ-MnO2 nanosheet assemblies with 150 m2 g−1 specific surface area are prepared by exfoliation of crystalline KxMnO2 and subsequent reassembly. Equilibration at different pH introduces intentional Mn vacancies into the nanosheets, increasing pseudocapacitance to over 300 F g−1, reducing charge transfer resistance as low as 3 Ω, and providing a 50% improvement in cycling stability. X-ray absorption spectroscopy and high-energy X-ray scattering demonstrate a correlation between the defect content and the improved electrochemical performance. The results show that Mn vacancies provide ion intercalation sites which concurrently improve specific capacitance, charge transfer resistance and cycling stability.
Similar content being viewed by others


Introduction
Manganese dioxide (MnO2) in its many forms has been the subject of much study for electrochemical capacitor applications1,2. In general, supercapacitors can be classified into two types: (i) electrical double-layer capacitors, which depend on charge separation at the electrode/electrolyte interface without Faradic process; and (ii) pseudocapacitors, which depend on Faradic redox reactions3,4,5. ‘Extrinsic’ pseudocapacitance has recently emerged as a subclassification of materials that host ion intercalation but are engineered to short length scales to reduce diffusion distances such that the discharge behaviour becomes linear and no structural phase changes occur6,7.
The birnessite form of MnO2 (δ-MnO2), comprising stacked sheets of edge-shared MnO6 octahedra with interlayer alkali ions8,9,10, has been studied for some time and shows both double-layer and Faradaic charge storage1,11,12. Two-dimensional (2D) δ-MnO2 generally exhibits improved capacitance and rate behaviour compared to other polymorphs because the interlayer galleries provide high-speed pathways for diffusion of protons or alkali cations during the charge and discharge processes13,14,15. However, the low electrical conductivity of δ-MnO2 (10−5 to 10−6 S cm−1) has greatly limited its application16, prompting study of composite electrodes containing graphene17,18, carbon nanotubes19,20, carbon fibres21,22 and so on. In addition, nanostructuring has been employed to improve the surface area and capacitance, for example by growing nanoneedles23, nanoflowers24, nanoparticles25 and so on.
In recent years, intentional creation of cation vacancies has been explored to increase the charge storage capacitance of transition metal oxides, where cation vacancies provide additional cation intercalation sites26. Cation vacancy content may be controlled via aliovalent cation substitution27, anion substitution28, reducing or oxidizing heat treatments29, or by equilibrating the oxides in pH-controlled suspensions30. The first studies correlating cation vacancies and charge storage properties were published by Ruetschi in the mid-1980s for intergrowth γ-MnO2 phases31,32,33. Metal vacancy content can be quite large in some cases, for example, Wei et al.28 modified anatase TiO2 with monovalent F− and OH− anions to form up to 22 at%  for additional Li storage. Similarly, Koo et al.34,35 transformed Fe3O4 (spinel) into hollow γ-Fe2O3 rods containing up to 44% vacant iron sites, and showed that Li and Na ion intercalation is possible without structural phase transformations.
for additional Li storage. Similarly, Koo et al.34,35 transformed Fe3O4 (spinel) into hollow γ-Fe2O3 rods containing up to 44% vacant iron sites, and showed that Li and Na ion intercalation is possible without structural phase transformations.
Little work on the formation of cation vacancies in δ-MnO2 nanosheets has been reported to date, but extensive study of the important role of birnessite-like MnO2 in photosynthesis, ion sorption and other bio- and geochemical processes provides rich literature on its behaviour in aqueous environments36. Most recently, Manceau et al.30, building from many earlier works on Mn oxidation state in K-birnessites10,37, systematically investigated the effects of pH on cation vacancy content in phyllomanganate nanoparticles. Their approach exploited high-energy X-ray scattering to perform pair distribution function (PDF) analysis, truncation rod analysis and simulations using the Debye equation to show that lower pH causes migration of Mn from the MnO2 nanosheet to the sheet surface. Thus, the authors were able to quantify Mn vacancies in the nanosheets and Mn3+ cations displaced to the interlayers. Later work by Marafatto and coworkers36 employed sub-picosecond optical and X-ray absorption spectroscopy to track the mechanisms of Mn reduction under illumination to quantify the effects of different interlayer cations on MnO2 photo-reduction rates. Their results support the Mn redox reaction mechanism proposed by Manceau et al.30, including displacement of the Mn3+ cation to the interlayer gallery.
Additional recent work has been focused on determining mechanisms of charge storage. For example, in situ X-ray absorption studies have been used to show when the Faradaic reactions occur in MnO2 nanosheets38 and to track the average Mn oxidation state across wide voltage swings39,40,41. Similarly, in situ Raman spectroscopy has been applied to track changes in the vibrational bands and has highlighted cation size effects over the Li, Na and K series42.
In the present work, we exfoliate and reassemble δ-MnO2 nanosheets to form 3D macroporous pseudocapacitive electrodes with controlled concentration of Mn point defects and Mn3+/4+ ratios. Electrochemical and high-energy X-ray scattering measurements provide direct evidence that intentional Mn ion defects and Mn reduction synergistically improve supercapacitor performance. The three-dimensional (3D) assembly and defect control represent straightforward and industrially scalable approaches to improving specific capacitance.
Results
Phases and microstructure
The morphologies of pristine KxMnO2, and its protonated form, HxMnO2, are shown in Supplementary Fig. 3. From Supplementary Fig. 3a,b it can be seen that the KxMnO2 and HxMnO2 particles are platy, with lateral dimensions in the range of one micron, and that proton exchange does not alter the grain morphology. High-magnification SEM and TEM images are shown in Fig. 1a–c for an individual nanosheet (Fig. 1a,b) and the reassembled MnO2 nanostructure (Fig. 1c). The images demonstrate that the sheets are generally flat, with some scrolling at the edges as has been noted in other nanosheet studies43,44. The atomic force microscopy (AFM) image in Supplementary Fig. 4 also displays nanosheet fragments with flat surfaces and thickness of five nanosheets, suggesting that some of the nanosheets exfoliate into bunches or restack to a small extent upon drying. The flocculated nanosheet samples exhibit 3D porous nanostructures (Fig. 1d; Supplementary Fig. 3c) and equilibration at different pH values have no influence on their morphologies. The observation of porous structures after reassembly highlights the usefulness of our ultrasonic-assisted exfoliation and flocculation procedure, where the exfoliation rate is greatly enhanced compared with other reported procedures45,46.

(a) SEM image and (b) bright-field TEM image of exfoliated MnO2 nanosheets, (c) high-magnification SEM image and (d) SEM image of reassembled MnO2 nanostructures treated in pH=2 solution for 24 h. Scale bar, 50 nm (a–c); 500 nm (d).
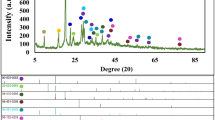
Synchrotron diffraction data for the parent KxMnO2 is shown in Supplementary Fig. 5. Rietveld refinement reveals that the as-synthesized parent material is layered birnessite, primarily exhibiting the monoclinic polytype with less than 10 wt% of the rhombohedral polytype and no additional crystalline phases. The X-ray diffraction patterns (Fig. 2) for protonated and reassembled MnO2 demonstrate that phase purity was achieved during synthesis and that exfoliation and reassembly yields complex nanostructures as evidenced by peak broadening and asymmetry. After reassembly of the nanosheets, in-plane hk0 reflections remain discernible, indicating that the crystalline nature of the 2D sheets is preserved. Further, the derived PDF is consistent both with literature47 and the calculated PDF of a single δ-MnO2 nanosheet (Supplementary Fig. 6), further confirming the δ-MnO2 nanosheet motif is maintained. The broadening of the 00l basal reflections shows that although some sheet-to-sheet restacking occurs, the stacks are on the order of only 3–4 nm (estimated using the Scherrer equation), which is consistent with the AFM results. Shifts of the basal reflections result from increased water content in the reassembled nanosheet floccules compared with the proton-exchanged form.

XRD patterns of (a) protonated MnO2, (b) reassembled MnO2 treated in pH=4 solution for 24 h, (c) reassembled MnO2 treated in pH=2 solution for 24 h. Data collected on APS 11-ID-B.
Nitrogen adsorption–desorption isotherms were used to quantify the specific surface area (SSA) of all specimens as shown in Fig. 3. While protonated MnO2 showed no evidence of mesopores, the reassembled MnO2 nanostructures show typical type IV isotherms (IUPAC classification) with distinct H3-type hysteresis loops, a result of open slit-like mesopores48. The Brunauer Emmet and Teller (BET) SSA were 120±0.4 m2 g−1 for the pH=2 sample and 144±1 m2 g−1 for the pH=4 sample, and only 4.5±0.1 m2 g−1 for HxMnO2, where the former values are roughly double the surface areas reported by Song et al.49 for reassembled oxide nanosheets. Altogether, the XRD, BET and microscopy show that the reassembled nanosheets have macro- and mesopores, with the mesoporosity arising due to loose agglomeration of randomly oriented sheet clusters. The extent of exfoliation and/or restacking, typical sheet thicknesses determined by AFM and average crystallinity in the sheet stacking direction as determined by XRD are similar or better for our MnO2 specimens than those reported recently for MnO2, TiO2, Co3O4, ZnO and WO3, for example44. Therefore, the typical structures in Fig. 1d are of the form of edge-to-face assembled nanosheet booklets, with ‘wall thicknesses’ of up to 4 nm. The high SSA of the MnO2 nanosheet assemblies facilitates infiltration of the electrolyte into the porous electrode to enhance the specific capacitance6,50.

N2 adsorption–desorption isotherms of (a) protonated MnO2, (b) reassembled MnO2 treated in pH=2 solution for 24 h, (c) reassembled MnO2 treated in pH=4 solution for 24 h. Curves (b) and (c) are offset by 50 and 200 cm3 g−1 STP, respectively.
During soft chemical processing, the surface and interlayer tetrabutyl ammonium hydroxide (TBAOH) and water content may vary with processing conditions. The water content in protonated, pH=2 and pH=4 samples was obtained by thermogravimetric analysis (Supplementary Fig. 7). The presence of structural water enables rapid proton or alkali cation transport within the interlayer, which is beneficial for increasing the charge storage properties6. The slightly higher water content in the pH=4 versus pH=2 sample is a result of its slightly larger surface area. The presence of residual TBAOH in the reassembled samples was indicated by a small mass loss (∼3%, corresponding to 0.01 TBAOH per MnO2 formula unit and therefore about 9% surface coverage) in the temperature region of 200–400 °C (refs 51, 52). Infrared spectroscopy (Supplementary Fig. 8) and X-ray photoelectron spectroscopy (XPS) (Supplementary Fig. 9) were also used to detect remnant TBAOH in the nanosheet assemblies after processing. Infrared was used to show that the TBAOH is removed from the nanosheet assemblies after one charge–discharge cycle, as shown in Supplementary Fig. 10. Although it is not possible to completely remove the TBAOH molecules from the nanosheet assemblies by washing, its small surface coverage (∼9%) and easy extraction during the first electrochemical cycle together suggest that it has limited if any influence on the measured electrochemical performance.
Redox and defects
The oxidation state of the Mn ions in all samples was investigated using Mn K-edge X-ray absorption near-edge spectroscopy (XANES) and XPS. The XPS analysis is described in Supplementary Information (Supplementary Fig. 11). Figure 4a shows the edge spectra of standard materials including MnO, Mn3O4, Mn2O3 and MnO2. The XANES spectra of the HxMnO2, pH=2 and 4 nanosheet assemblies, and reference materials Mn2O3 and MnO2 are shown in Fig. 4b. The line profiles are characterized by features that correspond to a pre-edge range with two weak broad peaks at 6,540–6,545 eV, a main-edge range that has one inflection point A, and the resonance peak range B49,53. The weak pre-edge peaks P and P′ correspond to the dipole-forbidden 1s→3d transition54. All three samples exhibit higher intensity of peak P′ than for peak P, but the peak P′ is less intense as compared with the β-MnO2 reference. This observation confirms the existence of mixed oxidation states of Mn3+/Mn4+ in the samples. Also, the higher intensity ratio of P′ to P for HxMnO2 compared with the pH=2 and 4 samples indicates that it has less Mn3+, because the relative intensity of the peaks P′/P is proportional to the average oxidation state54.

X-ray absorption measurements of as-prepared three samples and reference Mn oxide materials. (a) XANES spectra of reference materials MnO, Mn3O4, Mn2O3 and MnO2. (b) XANES spectra of protonated MnO2, pH=2 and 4 treated reassembled MnO2. The reference materials of Mn2O3 and MnO2 from (a) are also shown for of ease comparison. (c) First derivative curves corresponding to the samples shown in b. (d) Average oxidation state of Mn for the samples and standards derived from the K-edge energy.
The main absorption range can be assigned to the dipole-allowed 1s→4p transition. The associated edge energy is usually taken as the energy of the peak in the first derivative, which corresponds to the inflection point of the main edge in the XANES spectra (Fig. 4c). Clearly, the main absorption edge (A) progressively shifts to lower energies with decreasing pH, implying lower pH progressively reduces Mn to the trivalent state. Also, the presence of the intense peak B for the nanosheet assemblies indicates that they are mainly comprised of edge-shared MnO6 octahedra55. This observation further confirms that the δ-MnO2 lattice is not dissolved by equilibration in HCl at pH as low as 1.
The average oxidation state (AOS) of Mn was determined by establishing a linear relationship between the K-edge energy and Mn oxidation state (Fig. 4d). The AOS of Mn is 3.59 for HxMnO2, 3.36 for the pH=4 MnO2 nanosheet assembly, and 3.24 for the pH=2 variant. These values are also listed in Table 1 for correlation with the PDF and electrochemical results discussed below. The dependence of Mn valence on pH generally follows that reported earlier by Manceau and coworkers30 for bulk Na-saturated δ-MnO2 (birnessite) powders, where the Mn AOS varied from 3.81 at pH=9 to 3.69 for pH=3. For the alkali-free MnO2 samples studied herein, we find that treating the exfoliated MnO2 assemblies at lower pH increases the Mn reduction to a greater extent than found for crystalline NaxMnO2 by Manceau and coworkers30. Furthermore, we find competing steric and thermodynamic effects by comparison of the proton-exchanged HxMnO2 to the exfoliated and reassembled variants. The AOS of HxMnO2 treated at pH<1 is 3.59, whereas the exfoliated samples treated at higher pH have AOS values of 3.36 (pH=4) and 3.24 (pH=2). We attribute the lesser extent of Mn reduction in crystalline HxMnO2 to steric hindrance by the interlayer galleries which are crowded with protons and water. This has the two-fold effect of hindering access of aqueous H3O+ to reducible Mn4+, and of preventing displacement of Mn3+ to the sheet surface. Indeed, earlier work by Gaillot et al.10 noted that during thermal reduction of  in crystalline K-birnessite, Mn vacancies were not formed despite the unfavourable in-sheet lattice strain due to Jahn–Teller distortions inherent to Mn3+. The interactions of neighbouring in-plane Mn3+ and Mn4+ sites therefore contributes both strain and electrostatic-driven components to the energetics of δ-MnO2 defect equilibration.
in crystalline K-birnessite, Mn vacancies were not formed despite the unfavourable in-sheet lattice strain due to Jahn–Teller distortions inherent to Mn3+. The interactions of neighbouring in-plane Mn3+ and Mn4+ sites therefore contributes both strain and electrostatic-driven components to the energetics of δ-MnO2 defect equilibration.
High-energy X-ray scattering and pair distribution function analysis were undertaken to further characterize the defects formed in soft chemically reduced MnO2 nanosheets, with the specific goal of correlating the quantity of Mn point defects with Mn reduction and electrochemical performance. PDF analysis probes not only the local atomic bonding motifs but also intermediate and long-range order, thus making it an appropriate tool for investigating the structures of poorly-crystalline materials and nanoparticles that yield diffraction patterns with large amounts of diffuse scattering47.
The observed scattered intensity, the reduced structure function F(Q) and the associated PDF for the reassembled nanosheets are shown in Fig. 5. We focus our attention on two PDF correlation peaks: the in-plane Mn–Mn peak at 2.89 Å and the Mn– distance at 3.45 Å (Fig. 6a). Two notable advantages of the PDF method are that these two Mn correlations are unique in the alkali-free δ-MnO2 structure (Fig. 6b) and that the integrated area of a PDF peak represents the number density of that specific correlation type56. We leverage these facts to obtain useful estimates of Mn vacancy concentrations in lieu of developing complete structure models, which are notably difficult for these material systems57,58.
distance at 3.45 Å (Fig. 6a). Two notable advantages of the PDF method are that these two Mn correlations are unique in the alkali-free δ-MnO2 structure (Fig. 6b) and that the integrated area of a PDF peak represents the number density of that specific correlation type56. We leverage these facts to obtain useful estimates of Mn vacancy concentrations in lieu of developing complete structure models, which are notably difficult for these material systems57,58.

(a) I(Q) and (b) F(Q) for the pH=2 and pH=4 treated MnO2 nanosheets assemblies, and (c) G(r) for the pH-treated samples, protonated MnO2 (HxMnO2), and parent phases (KxMnO2). The basal reflections evident in I(Q<2 Å−1) indicate a measure of restacking. The variation in intensity as a function of pH indicates a variation in the interlayer structure attributable to either surface water or Mn. The inverse trends in the Mn–Mn peak (2.89 Å) and Mn–MnIL peak (3.45 Å) as a function of pH support the Mn3+ displacement model recommended by Manceau et al.30

Using the resolved in-plane Mn-Mn correlation peak (a) we can empirically estimate the Mn surface Frenkel (circled) concentration using a Gaussian peak and linear baseline (b). The PDF amplitude is normalized to the Mn–O correlation peak; therefore the ratio of nanosheet Mn-Mn to KxMnO2 Mn-Mn peak areas represents the fractional Mn-occupancy of the nanosheet assembly. δ-MnO2 equilibrated at pH=2 is shown here.
To compare the PDF with respect to different samples, we have normalized our data to the amplitude of the 1.9 Å Mn–O peak. This choice is based on the implicit assumption that all Mn ions are coordinated in a six-fold environment. We further assume that the intrinsic  population in the parent phase KxMnO2 is on the order of parts per million, which will be negligible compared to the per cent-level vacancies in the MnO2 nanostructures. Consequently, the relative in-plane Mn concentration can be calculated according to the following equation:
population in the parent phase KxMnO2 is on the order of parts per million, which will be negligible compared to the per cent-level vacancies in the MnO2 nanostructures. Consequently, the relative in-plane Mn concentration can be calculated according to the following equation:

Where the superscript 0 denotes quantities in KxMnO2, the subscript i denotes quantities for a derivative sample, and [Mn]0 is taken as 1. The absolute accuracy of the values obtained using equation (1) will be influenced by the correlations between interlayer water or hydroxyl pairs, which perturb the low-r PDF region, leading to overestimation of the  content47. While there is notable difficulty in absolute quantification of Mn defects in these materials, this method should provide reasonable relative quantification in the derived nanosheet assemblies. Although estimation of error is not possible with this empirical approach, a per cent level of uncertainty is expected for the calculated Mn concentrations.
content47. While there is notable difficulty in absolute quantification of Mn defects in these materials, this method should provide reasonable relative quantification in the derived nanosheet assemblies. Although estimation of error is not possible with this empirical approach, a per cent level of uncertainty is expected for the calculated Mn concentrations.
As shown in Fig. 6a, the  concentration, which is equivalent to the
concentration, which is equivalent to the  concentration, is then is 1−[Mn]i in fractional units. Here we define this type of defect as a ‘surface Frenkel’ defect, where displacement of the in-plane Mn to the nanosheet surface is reminiscent of the Frenkel defect vacancy-interstitial pair. The results of our analysis are summarized in Table 2. Consistent with the model of Manceau et al.30, we observe an increase in
concentration, is then is 1−[Mn]i in fractional units. Here we define this type of defect as a ‘surface Frenkel’ defect, where displacement of the in-plane Mn to the nanosheet surface is reminiscent of the Frenkel defect vacancy-interstitial pair. The results of our analysis are summarized in Table 2. Consistent with the model of Manceau et al.30, we observe an increase in  with decreasing pH, as well as the appearance of a PDF peak at a distance not found in the δ-MnO2 structure, which corresponds to
with decreasing pH, as well as the appearance of a PDF peak at a distance not found in the δ-MnO2 structure, which corresponds to  . The increase in concentration of surface Frenkel defects by ∼30% between the pH=4 and pH=2 samples supports the hypothesis that increased proton sorption at the MnO2 surface in more acidic electrolytes expels more in-plane Mn leading to the formation of more Mn vacancies.
. The increase in concentration of surface Frenkel defects by ∼30% between the pH=4 and pH=2 samples supports the hypothesis that increased proton sorption at the MnO2 surface in more acidic electrolytes expels more in-plane Mn leading to the formation of more Mn vacancies.
As noted for the Mn valence, the PDF analysis likewise shows an apparent contradiction in the surface Frenkel defect content of the crystalline HxMnO2 when compared to the defect content of the pH=2 and 4 samples. As shown in Table 1, while the protonated form HxMnO2 is equilibrated at pH<1, its defect concentration (18.3%) is smaller than that in the pH=2 sample (26.5%) and is even smaller than that of the pH=4 sample (19.9%). As noted earlier, previous work shows that Mn3+, with its Jahn–Teller distortion, may be accommodated in MnO2 sheets in crystalline birnessites59. Table 1 shows that ∼2/3, 1/2 and 1/4 of the Mn ions remaining in the nanosheets are reduced to Mn3+ for the pH=2, 4 and HxMnO2 samples, respectively. The PDF and XANES analyses are therefore complementary in demonstrating that surface Frenkel defects and Mn reduction are more favourable in high-surface area nanosheet assemblies, with steric limitations reducing the extent of the reactions in well-crystalline birnessites.
Electrochemical measurements
As shown in Fig. 7a, the cyclic voltammetry (CV) curves for all three samples exhibit largely rectangular shapes in the potential window from 0 to 1 V, which indicates capacitive behaviour. The absence of clear redox peaks for protonated and pH=4 samples implies that the electrodes are charged and discharged at a pseudo-constant rate over the whole voltammetric cycle. However, the presence of broad redox peaks for the pH=2 sample indicates that ion intercalation is an active and detectable charge storage mechanism for this specimen. Indeed, even broader redox peaks are noted for the pH=3 sample (Supplementary Fig. 12a), while no redox peaks are found for the pH=4 and pH=9 (Supplementary Fig. 12b) samples. The marked increase in Mn surface Frenkel defects with decreasing pH appears to enhance the intercalation reaction. This is also reflected in Fig. 7b–d, which shows the galvanostatic charge–discharge curves for the samples between 0 and 1 V under different current densities. The potential does not linearly change with time, a behaviour typical for pseudocapacitive materials that involve redox reactions.

Electrochemical testing results: (a) CV curves for the samples at 50 mV s−1 scan rate; (b), (c) and (d) galvanostatic charge–discharge curves of the pH=2, 4 and HxMnO2 samples; (e) comparison of specific capacitance for the samples as a function of current density; and (f) cycle stability at constant current density of 5 A g−1 between 0 and 1 V.
The specific capacitance of the electrode measured by the galvanostatic discharge method can be calculated as:

where C (F g−1) is the specific capacitance, I (A) is the constant discharge current, Δt (s) is the discharge time, ΔV (V) is the potential window, and m (g) is the mass of the active material in the electrode. The specific capacitances of the three samples obtained at different current densities are summarized in Fig. 7e. At 0.2 A g−1, the specific capacitance is 306 F g−1 for the pH=2 sample, 209 F g−1 for the pH=4 sample and 103 F g−1 for crystalline HxMnO2. With increasing current density, the specific capacitance decreases gradually for all the three samples.
The defect chemistry also has an impact on cycling stability, demonstrated using the pH=2 and 4 nanosheet assemblies, which is shown in Fig. 7f. After 1,000 charge–discharge cycles using a very high-current density of 5 A g−1 (2.5 mA cm−2), the pH=2 sample shows good stability, retaining 83% of its initial specific capacitance. In contrast, the pH=4 sample retains only 68% of its initial capacitance. Our ongoing work involves tracking the reversibility of the electrochemical strains induced during cycling, and although we have not yet uncovered the origins of the cycling fade, our results are promising when compared with earlier work, for example for MnO2 nanostructures which retain ∼92% of the initial capacity after 1,000 cycles when using a current density five times smaller49.
Electrochemical impedance spectroscopy (EIS) was employed to measure the charge transfer resistance of each electrode with the results shown in Fig. 8a. All plots exhibit a straight line in the low-frequency region and a single semicircle in the high-frequency region, indicating a diffusion-limited step in the low frequency region and a charge transfer limited step in the high-frequency region60. The Nyquist plots were further modelled and interpreted by using an appropriate electrical equivalent circuit, which is shown in the inset in Fig. 8a. Rs is a combined resistance including ionic resistance of the electrolyte, intrinsic resistance of the substrate and contact resistance at the active material/current collector interface. Rct is the charge transfer resistance caused by the Faradaic reaction. Zw is the Warburg resistance which is related to ion diffusion in the electrolyte, CPE is a constant phase element and CL is the limit capacitance61,62,63. The calculated charge transfer resistances (Rct) extracted from the high-frequency range were 23, 15 and 3 Ω for the HxMnO2, pH=4 and pH=2 samples, respectively, as shown in Fig. 8b.

(a) Nyquist plots of the samples in the frequency range of 0.1 Hz–100 kHz at an open circuit potential of 5 mV (inset shows the electrical equivalent circuit used for fitting the impedance spectra); and (b) comparison of the charge transfer resistance among the three samples (values obtained from the fitted data).
Table 1 summarizes the characteristics of the electrodes and their properties, showing several clear trends. First, the XPS and XANES studies, combined with quantification of surface Frenkel defects, demonstrate that the Mn3+/4+ ratio in the nanosheets trends with pH treatment. More specifically, Mn4+→Mn3+ reduction does not only result in formation of surface Frenkel defects but some of the Mn within the sheets is reduced to the trivalent state. As calculated based on the data in Table 1, ∼2/3, 1/2 and 1/4 of the Mn ions remaining in the nanosheets are reduced to Mn3+ for the pH=2, 4 and HxMnO2 samples, respectively.
The specific capacitance for the reassembled nanosheets correlates with the surface Frenkel defect population and Mn3+ content, as well as charge transfer resistance. For example, the surface areas of the pH=2 and 4 samples are similar, but the sample treated at pH=2 has 50% higher capacitance, 47% more Na+ ion intercalation, × 5 smaller charge transfer resistance, with 33% more surface Frenkel defects, highlighting the importance of the cation defects on Na+ ion intercalation.
The surface Frenkel defect provides two likely intercalation sites: the Mn ion vacancy, which is accessible from one side of the nanosheet; and the undercoordinated MnO6 surface octahedron on the opposite side of the nanosheet (Fig. 6a). In parallel, the defect reaction provides a large concentration of Mn3+ cations, which can presumably participate in polaron hopping conduction, thus improving electrical conductivity and charge transfer efficiency64,65. The specific mechanism of Na+ ion intercalation into the nanosheets is a topic of current study in our group, but we speculate that there exists some synergistic effect of the defect content and Mn redox that together define the charge transfer resistance and specific capacitance. The electrochemical cycling behaviours suggest that is likely that steric effects such as local distortions in the nanosheets around vacancies are active in relieving electrochemical strains that occur during cycling, and thus define to a large extent the specific capacitance.
Discussion
Electrostatic assembly of δ-MnO2 nanosheets in suspension under carefully controlled experimental conditions yields self-assembled 3D porous nanostructures with surface areas of ∼150 m2 g−1. Equilibrating the reassembled nanosheets in varied pH controls the extent of Mn4+→Mn3+ reduction, as well as creating charged defect pairs we term ‘surface Frenkel defects’ comprising a Mn vacancy within the sheet and a six-fold coordinated Mn3+ site on the surface of the nanosheet. The Mn surface Frenkel defect content reaches 26.5% for the nanosheet assemblies equilibrated at pH=2 and 19.9% for the pH=4 sample. The XPS and XANES data indicate an increase of the Mn3+/Mn4+ ratio with decreasing pH and the electrochemical results show direct correlation of Mn cation defects with specific capacitance. The specific capacitance increased from about 200 F g−1 (pH=4) to over 300 F g−1 (pH=2) by intentional introduction of ∼30% surface Frenkel defects, while at the same time the charge transfer resistance decreased from ∼15 Ω to ∼3 Ω. Therefore, it is now clear that Mn surface Frenkel defects in δ-MnO2 nanosheets increase Na+ ion intercalation by providing new, low energy intercalation sites.
Methods
Chemicals and reagents
MnCO3, K2CO3, Na2SO4, acetylene black, poly (vinylidene fluoride) (PVDF) and nickel foil were purchased from Alfa Aesar. 6N hydrochloric acid (HCl) solution and sodium hydroxide (NaOH) were obtained from Fisher Scientific. The tetrabutylammonium hydroxide solution (TBAOH, 40 wt% in H2O) and N-methyl-2-pyrrolidone (NMP) were purchased from Sigma-Aldrich. All reagents were used as received without further purification.
Fabrication of δ-MnO2 nanostructures
Powder synthesis included mixing MnCO3 and K2CO3 powders in a molar ratio of 40 : 9, by milling in isopropyl alcohol for 10 min using a McCrone Micronizing Mill (McCrone Group, USA) with alumina media. The resulting suspension was dried on a hot plate for 30 min at 60 °C and then calcined in alumina crucible at 800 °C for 24 h in air. 0.5 g of the resulting layered KxMnO2 was proton ion-exchanged in HCl solution (1 mol l−1, 45 ml) in an ultrasonic bath at room temperature for 4 h. The ion exchange process was repeated two additional times, followed by washing with DI water and air drying. The absence of K ions was confirmed by energy dispersive spectroscopy and XPS survey scans shown in Supplementary Figs 1 and 2. In addition, the XPS survey scans showed a complete absence of any signal from Al ions, giving good confidence that no contamination by the Al2O3 milling media was encountered.
To obtain exfoliated MnO2 nanosheets, 0.35 g HxMnO2 was equilibrated with 32.5 ml aqueous TBAOH solution (30 ml H2O+2.5 ml 40 wt% TBAOH) for 4 h in an ultrasonic bath at room temperature. The resulting suspension was centrifuged at 10,000 r.p.m. for 10 min. to separate the remaining bulk HxMnO2 from the nanosheet suspension. Self-assembly of the nanosheets in the colloidal suspension was achieved by adding 6N HCl solution to the suspension at a constant rate of 1 ml min−1, while stirring to reach pH=2, resulting in flocculation to form high-surface area 3D porous structures.
Mn defects and redox were controlled by equilibrating the assembled nanosheets at target pH values by increasing the pH from 2 upwards using 1 mol l−1 NaOH additions and stirring for 24 h. Dry nanosheet assemblies were finally obtained by washing, centrifugation, rinsing in 2-propanol and drying overnight at room temperature. Thus, 3D porous MnO2 nanostructures assembled from ultrathin 2D δ-MnO2 nanosheets with controlled defect content were obtained. For comparison with the 3D assembled nanosheets, protonated bulk HxMnO2 without exfoliation was also electrochemically tested.
Characterization of the samples
Microstructures were studied using scanning electron microscopy (SEM, FEI Quanta 200) at 20 kV. Transmission electron microscopy and high-resolution SEM were carried out using a Hitachi HF-3300 TEM/STEM. The STEM unit has a secondary-electron detector, which allows simultaneous high-resolution SEM and TEM imaging at 300 kV. The thickness and crystallite dimension of the exfoliated MnO2 nanosheets were probed by multimode atomic force microscope (AFM, Bruker Dimension Icon) in tapping mode using antimony doped silicon tips. The exfoliated nanosheets were electrostatically attached to a clean silicon wafer by drying the nanosheet suspension, and imaged in air. Thermogravimetric analysis (TGA) was performed using a TA Instruments SQT-Q600 DTA/TGA under flowing air with a heating rate of 10 °C min−1. The presence of any functional groups from organic compounds was probed with infrared spectroscopy (Nicolet 6700 FT-IR Spectrometer, USA). The specific surface area and porosity were examined by nitrogen adsorption and desorption isotherms collected at 77 K using a Micromeritics TriStar II 3020 system. The local chemical environment of the samples was characterized using a PHI Quantera X-ray photoelectron spectrometer equipped with Al Kα radiation. Mn K-edge X-ray absorption near-edge spectroscopy measurements were carried out at the bending magnet beamline F3 at the Cornell High Energy Synchrotron Source (CHESS). Data were collected at room temperature in fluorescence mode using a Hitachi vortex 4-element silicon drift detector. All spectra were calibrated using the spectrum of Mn metal foil, and the software package ATHENA was used for analysis66. The Mn vacancy content in δ-MnO2 samples was determined using high-energy X-ray scattering and PDF analysis, with data collected at the Advanced Photon Source on beam-line 11-ID-B using the rapid-acquisition PDF geometry67. Data sets were collected using standard 1 mm Kapton capillaries in Debye-Scherrer geometry, Si<311> monochromated primary beam at 58.66 keV, and a silicon flat plate area detector. Scattering data for PDF extraction were collected over a Q-range of 0.4–24.5 Å−1, and the powder diffraction data for all samples was collected over a Q-range of 0.2–9.0 Å−1. 2D X-ray diffraction data were integrated to 1D using FIT2D68, after appropriately calibrating detector deviations from orthogonality and masking invalid pixels. CeO2 was used as the calibration standard for detector geometry. Meanwhile, CeO2 and Ni were used to evaluate the instrument response function. The PDF data was reduced using PDFgetX2 (ref. 69), which includes the appropriate corrections for inelastic scattering and energy-dependent detector response, in addition to experimental background and absorption corrections, amongst others.
Electrochemical measurements
The working electrode was prepared by mixing 80 wt% active material, 15 wt% acetylene black and 5 wt% PVDF in NMP solution. After stirring for 6 h, the homogeneous slurry was spread onto a Ni foil substrate with an area of 1 cm2, and then heated at 100 °C for 2 h to evaporate the solvent and obtain the electrode. The loading of the active material on the working electrode was in the range of 0.5–0.6 mg cm−2. The capacitive performance was measured using a CHI 650E electrochemical analyser (CHI, USA) with a conventional three-electrode cell. Ag/AgCl and platinum wire were used as the reference and auxiliary electrodes, respectively, with 1 M Na2SO4 aqueous electrolyte. Cyclic voltammetry scans were carried out from 0 to 1 V at a scan rate of 50 mV s−1. Galvanostatic charge–discharge was measured at different constant current densities from 0.2 to 10 A g−1. EIS was performed in the frequency range of 0.1 Hz–100 kHz at an open circuit potential of 5 mV. EIS data was fitted to an electrical equivalent circuit model using ZsimpWin (Version 3.21, EChem Software) software.
Data availability
The data that support the findings of this study are available from the authors on request.
Additional information
How to cite this article: Gao, P. et al. The critical role of point defects in improving the specific capacitance of δ-MnO2 nanosheets. Nat. Commun. 8, 14559 doi: 10.1038/ncomms14559 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ghodbane, O., Ataherian, F., Wu, N.-L. & Favier, F. In situ crystallographic investigations of charge storage mechanisms in MnO2 -based electrochemical capacitors. J. Power Sources 206, 454–462 (2012).
Wei, W., Cui, X., Chen, W. & Ivey, D. G. Phase-controlled synthesis of MnO2 nanocrystals by anodic electrodeposition: implications for high-rate capability electrochemical supercapacitors. J. Phys. Chem. C 112, 15075–15083 (2008).
Chen, S., Zhu, J., Wu, X., Han, Q. & Wang, X. Graphene oxide-MnO2 nanocomposites for supercapacitors. ACS Nano 4, 2822–2830 (2010).
Meng, C., Liu, C., Chen, L., Hu, C. & Fan, S. Highly flexible and all-solid-state paperlike polymer supercapacitors. Nano Lett. 10, 4025–4031 (2010).
Oh, S. & Kim, K. Synthesis of a new mesoporous carbon and its application to electrochemical double-layer capacitors. Chem. Commun. 1999, 2177–2178 (1999).
Augustyn, V., Simon, P. & Dunn, B. Pseudocapacitive oxide materials for high-rate electrochemical energy storage. Energy Environ. Sci. 7, 1597–1614 (2014).
Augustyn, V. et al. High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance. Nat. Mater. 12, 518–522 (2013).
Zhu, H. et al. Birnessite-type MnO2 nanowalls and their magnetic properties. J. Phys. Chem. C 112, 17089–17094 (2008).
Ma, R., Bando, Y., Zhang, L. & Sasaki, T. Layered MnO2 nanobelts: hydrothermal synthesis and electrochemical measurements. Adv. Mater. 16, 918–922 (2004).
Gaillot, A.-C., Lanson, B. & Drits, V. A. Structure of birnessite obtained from decomposition of permanganate under soft hydrothermal conditions. 1. Chemical and structural evolution as a function of temperature. Chem. Mater. 17, 2959–2975 (2005).
Pan, H.-A. et al. Investigating mechanisms underlying elevated-temperature-induced capacity fading of aqueous MnO2 polymorph supercapacitors: cryptomelane and birnessite. J. Electrochem. Soc. 162, A5106–A5114 (2015).
Brousse, T., Bélanger, D. & Long, J. W. To be or not to be pseudocapacitive? J. Electrochem. Soc. 162, A5185–A5189 (2015).
Vargas, O., Caballero, A., Hernán, L. & Morales, J. Improved capacitive properties of layered manganese dioxide grown as nanowires. J. Power Sources 196, 3350–3354 (2011).
Radhiyah, A. et al. Doubling of electrochemical parameters via the pre-intercalation of Na+ in layered MnO2 nanoflakes compared to α-MnO2 nanorods. RSC Adv. 5, 9667–9673 (2015).
Komaba, S., Ogata, A. & Tsuchikawa, T. Enhanced supercapacitive behaviors of birnessite. Electrochem. Commun. 10, 1435–1437 (2008).
Zhang, X., Yang, W. & Ma, Y. Synthesis of polypyrrole-intercalated layered manganese oxide nanocomposite by a delamination/reassembling method and its electrochemical capacitance performance. Electrochem. Solid-State Lett. 12, A95–A98 (2009).
Peng, L. et al. Ultrathin two-dimensional MnO2/graphene hybrid nanostructures for high-performance, flexible planar supercapacitors. Nano Lett. 13, 2151–2157 (2013).
Zhu, J. & He, J. Facile synthesis of graphene-wrapped honeycomb MnO2 nanospheres and their application in supercapacitors. ACS Appl. Mater. Interfaces 4, 1770–1776 (2012).
Hu, L. et al. Symmetrical MnO2–carbon nanotube–textile nanostructures for wearable pseudocapacitors with high mass loading. ACS Nano 5, 8904–8913 (2011).
Lee, S. W., Kim, J., Chen, S., Hammond, P. T. & Shao-Horn, Y. Carbon nanotube/manganese oxide ultrathin film electrodes for electrochemical capacitors. ACS Nano 4, 3889–3896 (2010).
Luo, Y. et al. Self-assembly of well-ordered whisker-like manganese oxide arrays on carbon fiber paper and its application as electrode material for supercapacitors. J. Mater. Chem. 22, 8634–8640 (2012).
Chen, Y.-C. et al. Highly flexible supercapacitors with manganese oxide nanosheet/carbon cloth electrode. Electrochim. Acta 56, 7124–7130 (2011).
Kong, D. et al. Three-dimensional Co3O4@MnO2 hierarchical nanoneedle arrays: morphology control and electrochemical energy storage. Adv. Funct. Mater. 24, 3815–3826 (2014).
Zhao, S. et al. Controlled synthesis of hierarchical birnessite-type MnO2 nanoflowers for supercapacitor applications. Appl. Surf. Sci. 356, 259–265 (2015).
Jiang, R., Huang, T., Liu, J., Zhuang, J. & Yu, A. A novel method to prepare nanostructured manganese dioxide and its electrochemical properties as a supercapacitor electrode. Electrochim. Acta 54, 3047–3052 (2009).
Rolison, D. R. & Nazar, L. F. Electrochemical energy storage to power the 21st century. MRS Bull. 36, 486–493 (2011).
Hahn, B. P. et al. Electrochemical Li-ion storage in defect spinel iron oxides: the critical role of cation vacancies. Energy Environ. Sci. 4, 1495–1502 (2011).
Li, W. et al. High substitution rate in TiO2 anatase nanoparticles with cationic vacancies for fast lithium storage. Chem. Mater. 27, 5014–5019 (2015).
Swider-Lyons, K. E., Love, C. T. & Rolison, D. R. Improved lithium capacity of defective V2O5 materials. Solid State Ionics 152, 99–104 (2002).
Manceau, A. et al. Short-range and long-range order of phyllomanganate nanoparticles determined using high-energy X-ray scattering. J. Appl. Crystallogr. 46, 193–209 (2013).
Ruetschi, P. Cation-vacancy model for MnO2 . J. Electrochem. Soc. 131, 2737–2744 (1984).
Ruetschi, P. Influence of cation vacancies on the electrode potential of MnO2 . J. Electrochem. Soc. 135, 2657–2663 (1988).
Ruetschi, P. & Giovanoli, R. Cation vacancies in MnO2 and their influence on electrochemical reactivity. J. Electrochem. Soc. 135, 2663–2669 (1988).
Koo, B. et al. Intercalation of sodium ions into hollow iron oxide nanoparticles. Chem. Mater. 25, 245–252 (2013).
Koo, B. et al. Hollow iron oxide nanoparticles for application in lithium ion batteries. Nano Lett. 12, 2429–2435 (2012).
Marafatto, F. F. et al. Rate and mechanism of the photoreduction of birnessite (MnO2) nanosheets. Proc Natl. Acad. Sci. USA 112, 4600–4605 (2015).
Gaillot, A. C., Drits, V. A., Manceau, A. & Lanson, B. Structure of the synthetic K-rich phyllomanganate birnessite obtained by high-temperature decomposition of KMnO4. Substructures of K-rich birnessite from 1,000 °C experiment. Micropor. Mesopor. Mater. 98, 267–282 (2007).
Yeager, M. et al. Highly efficient K0.15MnO2 birnessite nanosheets for stable pseudocapacitive cathodes. J. Phys. Chem. C 116, 20173–20181 (2012).
Nam, K.-W., Kim, M. G. & Kim, K.-B. In situ Mn K-edge X-ray absorption spectroscopy studies of electrodeposited manganese oxide films for electrochemical capacitors. J. Phys. Chem. C 111, 749–758 (2007).
Ma, S.-B. et al. Electrochemical properties of manganese oxide coated onto carbon nanotubes for energy-storage applications. J. Power Sources 178, 483–489 (2008).
Weng, Y. T. et al. Spatially confined MnO2 nanostructure enabling consecutive reversible charge transfer from Mn(IV) to Mn(II) in a mixed pseudocapacitor-battery electrode. Adv. Energy Mater. 5, 1500772 (2015).
Chen, D. et al. Probing the charge storage mechanism of a pseudocapacitive MnO2 electrode using in operando Raman spectroscopy. Chem. Mater. 27, 6608–6619 (2015).
Ma, R., Bando, Y. & Sasaki, T. Directly rolling nanosheets into nanotubes. J. Phys. Chem. B 108, 2115–2119 (2004).
Sun, Z. et al. Generalized self-assembly of scalable two-dimensional transition metal oxide nanosheets. Nat. Commun. 5, 3813 (2014).
Omomo, Y., Sasaki, T., Wang, L. & Watanabe, M. Redoxable nanosheet crystallites of MnO2 derived via delamination of a layered manganese oxide. J. Am. Chem. Soc. 125, 3568–3575 (2003).
Liu, Z.-h., Ooi, K., Kanoh, H., Tang, W.-p. & Tomida, T. Swelling and delamination behaviors of birnessite-type manganese oxide by intercalation of tetraalkylammonium ions. Langmuir 16, 4154–4164 (2000).
Zhu, M. et al. Structural study of biotic and abiotic poorly-crystalline manganese oxides using atomic pair distribution function analysis. Geochim. Cosmochim. Acta 81, 39–55 (2012).
Paek, S.-M. et al. Exfoliation and reassembling route to mesoporous titania nanohybrids. Chem. Mater. 18, 1134–1140 (2006).
Song, M.-S. et al. Porously assembled 2D nanosheets of alkali metal manganese oxides with highly reversible pseudocapacitance behaviors. J. Phys. Chem. C 114, 22134–22140 (2010).
Simon, P., Gogotsi, Y. & Dunn, B. Where do batteries end and supercapacitors begin? Science 343, 1210–1211 (2014).
Lew, C. M. et al. Pure-silica-zeolite MFI and MEL low-dielectric-constant films with fluoro-organic functionalization. Adv. Funct. Mater. 18, 3454–3460 (2008).
Mao, L., Zhang, K., Chan, H. S. O. & Wu, J. Surfactant-stabilized graphene/polyaniline nanofiber composites for high performance supercapacitor electrode. J. Mater. Chem. 22, 80–85 (2012).
Kuo, S.-L. & Wu, N.-L. Investigation of pseudocapacitive charge-storage reaction of MnO2nH2O supercapacitors in aqueous electrolytes. J. Electrochem. Soc. 153, A1317–A1324 (2006).
Lee, Y. R., Kim, I. Y., Kim, T. W., Lee, J. M. & Hwang, S. J. Mixed colloidal suspensions of reduced graphene oxide and layered metal oxide nanosheets: useful precursors for the porous nanocomposites and hybrid films of graphene/metal oxide. Chem. Eur. J. 18, 2263–2271 (2012).
Hwang, S.-J. et al. Local crystal structure around manganese in new potassium-based nanocrystalline manganese oxyiodide. J. Phys. Chem. B 106, 4053–4060 (2002).
Egami, T. & Billinge, S. J. Underneath the Bragg Peaks: Structural Analysis of Complex Materials Elsevier (2003).
Metz, P. et al. X-ray and neutron total scattering analysis of Hy·(Bi0.2Ca0.55Sr0.25)(Ag0.25Na0.75)Nb3O10·xH2O perovskite nanosheet booklets with stacking disorder. Powder Diffr. 31, 126–134 (2016).
Wang, H.-W., Naguib, M., Page, K., Wesolowski, D. J. & Gogotsi, Y. Resolving the structure of Ti3C2Tx MXenes through multilevel structural modeling of the atomic pair distribution function. Chem. Mater. 28, 349–359 (2015).
Oku, M., Hirokawa, K. & Ikeda, S. X-ray photoelectron spectroscopy of manganese-oxygen systems. J. Electron. Spectrosc. Relat. Phenom. 7, 465–473 (1975).
Gao, P. & Liu, D. Facile synthesis of copper oxide nanostructures and their application in non-enzymatic hydrogen peroxide sensing. Sensors Actuators B: Chem. 208, 346–354 (2015).
Chang, J. et al. Asymmetric supercapacitors based on graphene/MnO2 nanospheres and graphene/MoO3 nanosheets with high energy density. Adv. Funct. Mater. 23, 5074–5083 (2013).
Fan, Z. et al. Asymmetric supercapacitors based on graphene/MnO2 and activated carbon nanofiber electrodes with high power and energy density. Adv. Funct. Mater. 21, 2366–2375 (2011).
Shao, Y., Wang, H., Zhang, Q. & Li, Y. High-performance flexible asymmetric supercapacitors based on 3D porous graphene/MnO2 nanorod and graphene/Ag hybrid thin-film electrodes. J. Mater. Chem. C 1, 1245–1251 (2013).
Huang, M. et al. Self-assembly of mesoporous nanotubes assembled from interwoven ultrathin birnessite-type MnO2 nanosheets for asymmetric supercapacitors. Sci. Rep. 4, 3878 (2014).
Wei, W., Cui, X., Chen, W. & Ivey, D. G. Manganese oxide-based materials as electrochemical supercapacitor electrodes. Chem. Soc. Rev. 40, 1697–1721 (2011).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Chupas, P. J., Qiu, X., Hanson, J. C., Lee, P. L. & Clare, P. Rapid-acquisition pair distribution function (RA-PDF) analysis. J. Appl. Crystallogr. 36, 1342–1347 (2003).
Hammersley, A., Svensson, S., Hanfland, M., Fitch, A. & Hausermann, D. Two-dimensional detector software: from real detector to idealised image or two-theta scan. High Pressure Res. 14, 235–248 (1996).
Qiu, X., Thompson, J. W. & Billinge, S. J. PDFgetX2: a GUI-driven program to obtain the pair distribution function from X-ray powder diffraction data. J. Appl. Crystallogr. 37, 678–678 (2004).
Wojdyr, M. Fityk : a general-purpose peak fitting program. J. Appl. Crystallogr. 43, 1126–1128 (2010).
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. DMR-1409102. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We are indebted to K. Chapman at the Advanced Photon Source for valuable guidance in high-energy X-ray measurements. XANES measurements were conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-1332208.
Author information
Authors and Affiliations
Contributions
S.T.M., D.L. and D.D.E. conceived the research. P.G. and T.H. synthesized the materials. P.G. performed the electrochemical measurements. P.M. and S.T.M. performed the X-ray scattering measurements. P.G. and R.H. performed the XANES measurements. Y.G. performed the XPS and AFM measurements. J.Y.H. performed the TEM measurements. P.G., P.M. and T.H. performed the material characterization. All authors discussed and analysed the data. P.G., P.M. and S.T.M. wrote the paper. All authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures, Supplementary Table and Supplementary References (PDF 1267 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gao, P., Metz, P., Hey, T. et al. The critical role of point defects in improving the specific capacitance of δ-MnO2 nanosheets. Nat Commun 8, 14559 (2017). https://doi.org/10.1038/ncomms14559
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms14559
This article is cited by
-
The role of stacking fault tetrahedra on void swelling in irradiated copper
Communications Materials (2024)
-
Unlocking supercapacitive energy storage potential: Catalyzing electrochemically inactive manganese oxides to active MnO2 via heterostructure reconstruction
Nano Research (2024)
-
Selection of Cu2+ for intercalation from the electronegativity perspective: Improving the cycle stability and rate performance of δ-MnO2 cathode material for aqueous zinc-ion batteries
Science China Materials (2023)
-
Novel Synthesis and Characterization of Flexible MnO2/CNT Composites Co-deposited on Graphite Paper as Supercapacitor Electrodes
Journal of Electronic Materials (2022)
-
Hydrothermal Synthesis, Characterization, and Electrochemical Properties of MnO2-Titanate Nanotubes (MnO2-TNTs)
Journal of Electronic Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
