
Abstract
The great majority of thyroid cancers are of the non-medullary type. Here we report findings from a genome-wide association study of non-medullary thyroid cancer, including in total 3,001 patients and 287,550 controls from five study groups of European descent. Our results yield five novel loci (all with Pcombined<3 × 10−8): 1q42.2 (rs12129938 in PCNXL2), 3q26.2 (rs6793295 a missense mutation in LRCC34 near TERC), 5q22.1 (rs73227498 between NREP and EPB41L4A), 10q24.33 (rs7902587 near OBFC1), and two independently associated variants at 15q22.33 (rs2289261 and rs56062135; both in SMAD3). We also confirm recently published association results from a Chinese study of a variant on 5p15.33 (rs2736100 near the TERT gene) and present a stronger association result for a moderately correlated variant (rs10069690; OR=1.20, P=3.2 × 10−7) based on our study of individuals of European ancestry. In combination, these results raise several opportunities for future studies of the pathogenesis of thyroid cancer.
Similar content being viewed by others

Introduction
Thyroid cancer is the most common malignancy of the endocrine system and its incidence in industrialized countries has been rising over the past few decades. At present the incidence rate of thyroid cancer is 7.4 and 22.0 per 100,000 in the United States (US) for males and females of European ancestry, respectively1. In Iceland, the incidence rate is 4.1 and 10.7 for males and females, respectively2.
Thyroid tumours are classified into four main histology groups: papillary (PTC), follicular (FTC), medullary (MTC) and undifferentiated or anaplastic thyroid carcinomas. The majority of all thyroid tumours are non-medullary; either PTC (80-85%) or FTC (10-15%)3,4.
Thyroid cancer has been shown to have one of the strongest genetic component of all cancers, and the effect has been shown to extend beyond the nuclear family5,6,7. In an attempt to discover sequence variants conferring risk of thyroid cancer we previously performed two thyroid cancer genome-wide association studies (GWASs) in which we discovered, and published8,9, five risk variants, located on 2q35, 9q22.33, 8p12 and 14q13.3. These variants have since been widely replicated by other study groups10,11,12,13,14.
The purpose of our current study was to continue our search for thyroid cancer risk variants by building on our two previous thyroid cancer GWASs. Hence, we have expanded our study group as well as the number of markers analysed. This expansion provided us with both additional marker density and statistical power, resulting in the discovery of five new thyroid cancer risk loci, located at: 1q42.2, 3q26.2, 5q22.1 10q24.33 and 15q22.33. These results highlight several new potential focus points for future thyroid cancer research studies.
Results
Imputation and GWAS data
To search for additional thyroid cancer risk loci, we reanalysed our Icelandic GWAS data set after having expanded it close to seven-fold in terms of total number of participants included in the study and approximately doubling the number of variants analysed from our previously reported study9. The increase in number of variants is based on whole genome sequencing (WGS) of 15,220 Icelanders to an average depth of 34.5 × (see Methods). The Icelandic non-medullary thyroid cancer GWAS results are based on 1,003 patients and 278,991 controls (see Supplementary Table 1). Apart from previously reported8,9 variants or their correlates, located on: 2q35, 8p12, 9q22.33 and 14q13.3, no variants met our genome-wide significance criteria (Supplementary Table 2). The threshold for genome-wide significance in the present study was corrected for multiple testing using a weighted Bonferroni procedure based on functional impact of classes of variants15 (GWAS significance thresholds range between 2.6 × 10−7 and 7.9 × 10−10 depending on functional annotations; see Methods).
We then conducted a meta-analysis including the Icelandic results and four additional case-control groups of European descent, with populations from Columbus, Ohio and Houston, Texas, in the United States (US), the Netherlands and Spain. The number of study subjects in the Columbus study group has been increased by over four-fold from the previously reported9, and the Houston study group has not been included in any of the previously published GWASs of thyroid cancer. All these study subjects were whole-genome analysed for the first time in the present study by genotyping them using the Illumina Quad array chip genotyping platform. For the Spanish and the two US-study groups we performed genome-wide imputation using the 1000 Genomes Phase 3 Project data. For the Dutch study group, the genome-wide imputation was done using The Genome of the Netherlands16 (GoNL) whole-genome sequencing data set (see Methods). In total, the non-Icelandic data set consists of 1,998 patients and 8,559 controls. Per-allele odds ratios and P values for all variants in each of the five study group GWAS analyses were obtained using a logistic regression model. There was little evidence of systematic over dispersion of the test statistic (λGC=1.021–1.079; Supplementary Fig. 1). A fixed-effects meta-analysis was conducted for 7.1 million variants (with imputation info score≥0.90 in all study groups) in a total of 3,001 thyroid cancer patients and 287,550 controls.
Association with thyroid cancer
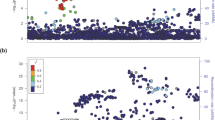
The strongest associations were with previously reported8,9 thyroid cancer risk variants or with variants in strong linkage disequilibrium (LD) with them, located on: 2q35, 8p12, 9q22.33 and 14q13.3 (Fig. 1, Table 1 and Supplementary Table 2). Only at the 2q35 locus was the variant reported in the present study (rs11693806) significantly stronger than the previously reported rs966423 (results for rs11693806[C] conditioned on rs966423: OR=1.31; P=9.5 × 10−8; whereas results for rs966423[C] conditioned on rs11693806 are: OR=1.03; P=0.61, see Supplementary Table 3). We also discovered five new regions, located on: 1q42.2, 3q26.2, 5q22.1, 10q24.33, 15q22.33, containing variants that are genome-wide significantly associated with thyroid cancer (Table 2 and Supplementary Fig. 2). The 15q22.33 locus contains two variants that associate with thyroid cancer independent of each other (see Table 3).

The Manhattan plot shows 7.1 million variants with high imputation information score (info≥0.90) from the thyroid cancer meta-analysis of GWAS data from 3,001 patients and 287,550 controls from five study groups of European descent coming from Iceland, the Netherlands, Spain and the USA. Shown are negative log10-transformed P values over 22 autosomes. Previously published risk loci are in black and risk loci reported in current study are in red.
Unlike the thyroid cancer risk variants previously reported8,9, none of the variants at these new risk loci is significantly associated (after correcting for multiple testing: P<0.01) with serum levels of thyroid-related hormones (that is, thyroid stimulating hormone (TSH), free-triiodothyronine (fT3) or free-thyroxine (fT4); see Supplementary Fig. 3 and Supplementary Table 4). Also, none of the newly discovered variants had a significant association with gender or age at diagnosis. However, the number of samples of the follicular subtype is too few to allow a meaningful comparison of the effect size for different histological subtypes of thyroid cancer.
Results for other previously reported risk variants
During the preparation of this manuscript, Ge et al.17 reported results from an association study of thyroid cancer in Chinese, analysing haplotype tagging variants located in the TERT-CLPTM1L region on 5p15.33; a region reported to contain several variants associated with risk of cancer in several organs. They found a significant association between rs2736100[C] and thyroid cancer in the Chinese. Results in the present study confirm this finding in populations of European ancestry (rs2736100[C]: OR=1.11; P=7.3 × 10−4, see Supplementary Table 3). However, in our study the strongest association results in this region were for rs10069690[T] (OR=1.20; P=3.2 × 10−7, see Table 2). Based on the 1000 Genome Project data set, the correlation between rs10069690 and rs2736100 is moderate in both individuals of European ancestry (r2=0.27) and Asians (r2=0.26). When we condition the two variants on each other, only results for rs10069690[T] remain significant (P=1.3 × 10−4 conditioned on rs2736100; compared to P=0.11 for rs2736100[C] conditioned on rs10069690, see Supplementary Table 3). Hence, it is likely that the association signals of these two variants reflect the same signal although rs10069690 seems to confer a slight refinement of it when moving from the Chinese to the European ancestry study groups.
For other previously reported18,19,20,21 GWAS-identified thyroid cancer risk variants, we nominally replicate results19 for rs13184587 located on 5q14.1 (OR=1.08, P=0.040; see Supplementary Data set 1).
Bioinformatics- and eQTL analyses of the novel risk variants
We next performed a bioinformatics- and eQTL (expression quantitative trait locus) analyses, searching for influences of these newly discovered variants on biologically functional elements or gene expression in normal thyroid tissue. Several variants yielded interesting results and had robust association with gene expression (see Supplementary Data set 2). The most interesting findings based on these analyses are discussed below.
At 1q42.2 the lead SNP rs12129938[A] (OR=1.32; P=4.0 × 10−11) lies within an enhancer in the first intron of PCNXL2. Little is known about the function of PCNXL2 but it is highly conserved and mainly expressed in the brain according to the GTEx Portal (accessed 12 June 2016: http://www.gtexportal.org/home/).
The second most significant variant, rs7902587[T] (OR=1.41; P=5.4 × 10−11), is located at 10q24.33, approximately16 Kb upstream of OBFC1. According to the HaploReg browser (accessed 1 June 2016: http://www.broadinstitute.org/mammals/haploreg/haploreg.php), rs7902587 is located in an enhancer region in fetal brain tissue. Several other variants, highly correlated (r2≥0.75) with rs7902587, have several hits within both promoters and enhancers in many different tissue types, including the thyroid. OBFC1 is a subunit of an alpha accessory factor (AAF) that stimulates the activity of DNA polymerase-alpha-primase, the enzyme that initiates DNA replication22. OBFC1 also appears to function in a telomere-associated protein complex (with C17ORF68 and TEN1 (ref. 23)). rs7902587 also correlates (r2=0.74) with variants reported24,25 to associate with longer telomeres as well as with a strongly correlated variant (r2=0.94) conferring risk of melanoma26.
rs73227498[A] (OR=1.37; P=3.0 × 10−10) at 5q22.1 is located in an intergenic region approximately 394 Kb upstream of NREP and 12 Kb downstream of EPB41L4A. According to GTEx, EPB41L4A has the second highest expression in thyroid of all tissues analysed (see Supplementary Fig. 4). However, the association of the thyroid cancer risk allele (rs73227498[A]) with expression of EPB41L4A (P=3.0 × 10−6) becomes non-significant when conditioned on the most significant cis-eQTL signal (rs821749) for EPB41L4A (P=0.29 for association of rs73227498 with expression of EPB41L4A when conditioned on rs821749). rs821749 is not genome-wide significantly associated with thyroid cancer (P=0.0015) in our study. Members of the EPB41L4A gene superfamily are thought to regulate the interaction between the cytoskeleton and plasma membrane. This locus also contains other variants strongly correlated (r2≥0.75) with rs73227498 that are located within regulator elements detected in several different tissues, including from the thyroid (see Supplementary Data set 2).
The 15q22.33 locus contains two independent association signals: rs2289261[C] with OR=1.23; P=3.1 × 10−9, and rs56062135[T] with OR=1.24; P=4.9 × 10−9 (see Table 3). The two variants are located intronic in SMAD3 that is an important transcriptional mediator of transforming growth factor-β (TGF-β) signalling. SMAD3 is most highly expressed in the thyroid of all tissues analysed according to GTEx (see Supplementary Fig. 5), and the thyroid cancer risk alleles of the two variants associate with increased expression of SMAD3 in normal thyroid tissue, (rs56062135[T] has β=0.46 s.d., P=4.8 × 10−14, and rs2289261[C] has β=0.29 s.d., P=1.6 × 10−6). According to GTEx, the association between rs56062135 and SMAD3 expression in thyroid tissue is the strongest cis-eQTL association signal reported for this gene (+/− 1 Mb around transcript start site). A conditional analysis of the expression data, adjusting the two variants for each other, demonstrates that both remain significant, although results for rs56062135 retain more of the significance (rs56062135[T], adjusted for rs2289261, had β=0.40 s.d., P=4.2 × 10−10, and rs2289261[C], adjusted for rs56062135, had β=0.14 s.d., P=0.021).
At 3q26.2, the lead variant, rs6793295[T] (OR=1.23; P=2.7 × 10−8), is a missense mutation (p.Ser249Gly) in LRRC34 and has (either itself or through a highly correlated variant) been previously reported to associate with several diseases and other traits, specifically: pulmonary fibrosis27, bladder cancer28, multiple myeloma29,30 and telomere length25,31,32. Little is known about the function of LRRC34 but its role has been suggested to be in ribosome biogenesis in pluripotent stem cells33. No significant association with expression of any gene is reported for rs6793295[T] in thyroid tissue, according to GTEx, but the strongest eQTL associations are with decreased expression of LRRC34 in transformed fibroblasts, tibial tissue and subcutaneous adipose tissue (β=−0.35 s.d., −0.39 s.d., and −0.28 s.d., respectively; P=1.5 × 10−8, 6.7 × 10−8 and 7.9 × 10−8, respectively). In addition to the lead variant, numerous variants, highly correlated (r2≥0.75) with rs6793295, are associated with thyroid cancer at this locus (see Supplementary Data set 2). Among them is another missense variant, rs10936600 (p. Leu286Ile), in LRRC34 and a splice-region variant rs9822885 in ACTRT3, a neighbouring gene. Another interesting gene located at this locus is TERC, which is the RNA component of the telomerase, involved in maintenance of telomeres. Variants, located at this region and strongly correlated (r2≥0.95) with the thyroid cancer risk variant, have been reported31,32 to associate with telomere length and, the allele conferring risk of thyroid cancer is correlated with the allele associated with longer telomeres.
As discussed above, rs10069690[T] (OR=1.20; P=3.2 × 10−7) is the strongest variant at 5p15.33 in our analysis. It is located in the fourth intron of the TERT gene and has previously been reported to confer risk of estrogen receptor negative breast- and ovarian cancer34. The authors of that study concluded that the cancer risk was not due to a telomere length association signal also reported by them for moderately correlated variants within this region (rs2736108 and rs7705526 have an r2 with rs10069690 of 0.04 and 0.34, respectively). Interestingly, the thyroid cancer risk allele of the variant (rs2736100[C]) reported in the Chinese study has also been found to associate with longer telomeres25 but that variant is only moderately correlated with the one reported in this region in our study (r2=0.27 between rs10069690 and rs2736100 in EUR) and therefore it is unclear if telomere length plays a role in conferring risk of thyroid cancer at this locus. Another study35 found the thyroid cancer risk allele of rs10069690[T] to be protective of prostate cancer and associated with increased expression of TERT in benign prostate tissue. According to GTEx, the expression level of TERT in normal thyroid tissue is too low to draw meaningful conclusion. Several other variants at this locus have been associated with numerous diseases, both benign and malignant according to the GWAS Catalog (The NHGRI-EBI Catalog of published GWASs (accessed on 21 June 2016): https://www.ebi.ac.uk/gwas/home).
Discussion
In summary, our meta-analysis has yielded five new thyroid cancer susceptibility loci. In addition, we confirm a risk locus recently reported17 in a study of Chinese thyroid cancer patients. Similarly, as with the bulk of previously reported thyroid cancer risk variants, the majority of these newly discovered variants are located either intronic or intergenic. Recently, the importance of this group of ‘noncoding’ variants has been coming clearer, as demonstrated for two of the previously discovered thyroid cancer risk variants, located intergenic at 9q22 and 14q13, and shown to affect the expression and events downstream of long non-coding RNA genes36,37,38. Interestingly, three of the variants reported here are located in regions reported to associate with telomere length and for two of them (rs6793295 on 3q26.2 and rs7902587 on 10q24.33) the allele associated with longer telomeres is correlated with the allele conferring risk of thyroid cancer. For the third variant (rs10069690 on 5p15.33) the correlation is weaker (r2=0.27 between rs10069690 and rs2736100) even though the direction of the association with thyroid cancer and telomere length is the same as for the other two variants. However, it remains to be determined what biological features account for involvement of these variants in the pathogenesis of non-medullary thyroid cancer.
Methods
Study populations
All Icelandic participants in this study are of European ancestry. Icelanders diagnosed with thyroid cancer were identified based on a nationwide list from the Icelandic Cancer Registry (ICR) (http://www.krabbameinsskra.is/indexen.jsp?icd=C73) that contains all Icelandic thyroid cancer patients diagnosed from 1 January 1955, to 31 December 2015. Thereof, 1,108 were non-medullary thyroid cancers. Out of the total list, genotypic information was available from 1,003 (71% are females) individual for the current study and their mean age at diagnosis is 53 years. The 278,991 Icelandic controls (49% females) used in this study consist of individuals belonging to different genetic research projects at deCODE. The controls had a mean age of 57 years. The controls were absent from the nationwide list of thyroid cancer patients according to the ICR. The study was approved by the Data Protection Commission of Iceland and the National Bioethics Committee of Iceland. Written informed consent was obtained from all subjects. Personal identifiers associated with medical information and blood samples were encrypted with a third-party encryption system as previously described39.
The Dutch study population consists of 85 non-medullary thyroid cancer cases (70% are females) and 4,956 cancer-free individuals (55% females). The cases were recruited from the Department of Endocrinology, Radboud University Medical Center (Radboudumc), Nijmegen, The Netherlands from November 2009 to June 2010. All patients were of self-reported European descent. Demographic and clinical characteristics were obtained from the patient's medical records. The average age at diagnosis of the patients was 40 years and average age of the controls was 55 years. The DNA for both the Dutch cases and controls was isolated from whole blood using standard methods. The controls were recruited within a project entitled ‘Nijmegen Biomedical Study’ (NBS). For a detailed description of this study, see Galesloot et al.40 The study was approved by the Ethical Committee and the Institutional Review Board of the Radboudumc, Nijmegen, The Netherlands and all study subjects gave written informed consent.
All subjects recruited in Columbus, USA, and included in the current study were of self-reported European descent and provided written informed consent. The 1,580 patients have a mean age of diagnosis of 43 years (74% are females) and were histologically confirmed papillary or follicular thyroid carcinoma (PTC) patients (including traditional PTC and follicular variant PTC). The 1,628 controls have a mean age of 45 years (74% are females) and are individuals without clinically diagnosed thyroid cancer from the central Ohio area. Genomic DNA was extracted from blood. The study was approved by the Institutional Review Board of the Ohio State University.
The study population recruited in Houston, USA, consisted of 250 papillary thyroid cancer (PTC) patients (69% are females) and 363 cancer-free controls (56% are females). The diagnosis of PTC was assigned by histologic review of the surgical or biopsy specimen, and the controls were recruited from among visitors to the institution. All subjects were of self-reported European descent. The mean age of diagnosis of PTC patients was 45 years and the mean age of controls at recruitment was 53 years. Genomic DNA was extracted from buffy coat cells isolated from peripheral blood. The study was approved by the MD Anderson Cancer Center Institutional Review Board and each subject provided written informed consent.
The Spanish study population consisted of 83 non-medullary thyroid cancer cases. The cases were recruited from the Oncology Department of Zaragoza Hospital in Zaragoza, Spain, from October 2006 to June 2007. All patients were of self-reported European descent. Clinical information including age at diagnosis, grade and stage was obtained from medical records. The average age at diagnosis for the patients was 49 years (71% are females). The 1,612 Spanish control individuals (51% are females) had a mean age of 47 and were approached at the University Hospital in Zaragoza, Spain, and were not known to have thyroid cancer. The DNA for both the Spanish cases and controls was isolated from whole blood using standard methods. Study protocols were approved by the Institutional Review Board of Zaragoza University Hospital. All subjects gave written informed consent.
Thyroid-related hormone measurements
TSH, fT4 and fT3 levels were measured in blood samples of Icelanders seeking medical care between the years 1997 and 2015 at the Landspitali University Hospital, the Clinical Laboratory in Mjodd, Reykjavik, Iceland, and Akureyri Hospital, Akureyri, Iceland. The QTL association results are based on measurements for the following number of Icelanders: TSH=188,057 individuals; fT3=51,047 individuals; fT4=120,879 individuals. The measurements were normalized to a standard normal distribution using quantile-quantile normalization and then adjusted for centre, gender, year of birth and age at measurement. For individuals for which more than one measurement was available we used the average of the adjusted normalized values. A generalized form of linear regression that accounts for the relatedness between individuals was used to test for the association of quantitative traits (that is, TSH, fT3 and fT4), with sequence variants.
Genotyping and statistical methods
The Icelandic thyroid cancer GWAS data set used in the current study is based on WGS, chip genotyping and long-range phasing of Icelandic population samples41. In brief, we whole genome sequenced 15,220 Icelanders using Illumina technology (Illumina, San Diego, CA, USA) to an average depth of at least 34 × , resulting in the identification of some 94 million variants. Using imputation assisted by long-range haplotype phasing42,43 and after removing variants with imputation information content below 0.8 as well as with an imputed minor allele frequency below 0.01%, we successfully inferred the genotypes of 32,463,443 variants in 434,571 Icelanders, of whom 151,677 had been genotyped using the Illumina chip genotyping platform. The remaining 282,894 Icelanders are first- and second-degree relatives of the chip-typed individuals and are imputed by aid of genealogic information. Of the patients used in this study 639 (64%) were chip genotyped and the remaining 364 were imputed by aid of genealogical information. Comparable numbers for the controls used are 126,251 (45%) chip genotyped and the remaining 152,739 were imputed by aid of genealogical information. In the meta-analysis only variants with imputation info score≥0.9 were included.
The Dutch, Spanish and US study samples were genotyped using Omni-1 Quad-bead chips (Illumina, San Diego, CA, USA). For each sample set, variants were excluded if they (i) had<94% yield, (ii) had<1% MAF, (iii) failed Hardy-Weinberg test (P<1 × 10−6) or (iv) showed significant (P<1 × 10−6) difference between genotype batches. Samples with<94% yield were excluded. The resulting genotypes were phased using SHAPEIT (v2.790) (ref. 44), and used to impute un-genotyped variants using IMPUTE2 (v2.3.2) (ref. 45). All but the Dutch samples were imputed using the 1000 Genomes Phase 3 reference data (October 2014 release) that includes phased genotypes for about 80 million variants and for 2,504 individuals of various ethnicities46. The Dutch study samples were imputed using the GoNL (ref. 16) data set generated by whole-genome sequencing of 249 Dutch trios (498 unrelated parents and 249 children).
Single track assay SNP genotyping
For validation purposes we directly genotyped the variants reported in Table 2 of the main text for a sub-set of 2,754 cases and controls form all five case-control groups of the present study. The correlation (r2) between imputed and directly genotyped was above 0.92 for all markers. Due to low imputation information score (ranging between 0.72 and 0.78) for s116909374 on 14q13.3 in the non-Icelandic study groups, this variant was directly genotyped in all available samples and the results combined with the Icelandic imputation results (which had imputation score=1.0) and presented in Table 1 and in Supplementary Table 2, The direct genotyping was carried out by deCODE Genetics in Reykjavik, Iceland, applying the Centaurus47 (Nanogen) platform.
Population structure
To study the population structure and the ancestry of samples in the Dutch, Spanish and US cohorts we used the ADMIXTURE (v 1.2) (ref. 48) and EIGENSOFT (v 6.0.1) (ref. 49) software. Samples were excluded if they were identified as ethnic outliers in the respective cohort, and to adjust for remaining population substructure ten principle components were included as covariates in the subsequent association analysis. Likewise, for the Icelandic cohort population, substructure was adjusted for by including county of origin as a covariate in the analysis.
Association testing
Logistic regression was used to test for association between variants and disease, assuming a multiplicative model, treating disease status as the response and expected genotype counts from imputation as covariates. For the Icelandic cohort this was done using software developed at deCODE genetics41, but the Dutch, Spanish and US cohorts were analysed using the SNPTEST (v.2.5) software50. Testing was performed using the likelihood ratio statistic. For the Icelandic study group patients and controls are matched on gender and age at diagnosis or age at inclusion but for the other study groups we did not adjust for these factors. Variants in the 1000 Genomes and GoNL imputation data sets were mapped to NCBI Build38 positions and matched to the variants in the Icelandic data set based on allele variation. Results from the different study groups were combined using a Mantel-Haenszel model51 in which the groups were allowed to have different population frequencies for alleles and genotypes but were assumed to have a common OR. Heterogeneity was tested by comparing the null hypothesis of the effect being the same in all populations to the alternative hypothesis of each population having a different effect using a likelihood ratio test. I2 lies between 0 and 100% and describes the proportion of total variation in study estimates that is due to heterogeneity.
Association significance thresholds
The threshold for genome-wide significant association in the current study was corrected for multiple testing using a class-specific Bonferroni procedure based on functional impact of classes of variants15. This yielded significance thresholds of: (i) 2.6 × 10−7 for 8,474 high-impact variants (comprised of: stop-gained, frameshift, splice acceptor or donor), (ii) 5.1 × 10−8 for 149,983 moderate-impact variants (comprised of: missense, splice-region variants and in-frame INDELs), (iii) 4.6 × 10−9 for 2,283,889 low impact variants (comprised of: synonymous variants 3′ and 5′ UTR variants), (iv) 2.3 × 10−9 for 3,913,058 intergenic and deep intronic variants overlapping DNase hypersensitivity sites and (v) 7.9 × 10−10 for 26,108,039 other variants (intergenic and deep intronic).
Genomic control adjustment
To account for inflation in test statistics due to cryptic relatedness and stratification within the case and control sample sets, we applied the method of LD score regression52. With a set of 1.1million variants we regressed the χ2 statistics from our GWAS scan against LD score and used the intercept as a correction factor. The LD scores were downloaded from an LD score database (accessed 23 June2015: ftp://atguftp.mgh.harvard.edu/brendan/1k_eur_r2_hm3snps_se_weights.RDS). The estimated correction factor was 1.079 for the Icelandic samples, mostly due to relatedness of the individuals; all P values in the Icelandic GWAS study were adjusted for this inflation. For the other study groups, the genomic adjustment factors were modest, in all cases below 1.043, and this was not adjusted for. For the combined meta-analysis, the correction factor was estimated to be 1.031.
Bioinformatics analysis
To inspect if the thyroid cancer risk variants might have regulatory effects or be located within regions with predicted biological features we performed an analysis selecting the lead variant at each of the newly discovered risk loci as well as all variants strongly correlated (r2≥0.75) with the lead variant. For this set of variants, we searched for overlaps with known regulatory regions as follows: first we used ENSEMBL to determine whether the variant had been assigned a regulatory region ENSR number. Then we examined the ENCODE data and looked for any evidence of ChIP-Seq transcription factor binding and DNaseI hypersensitivity sites53,54. We also looked for enhancer and promoter chromatin segmentation states using the 25-state HMM from the Roadmap consortium55. Then we looked for correlations between DNaseI hypersensitive sites and local gene expression using results described by Sheffield et al.56. Finally, we searched for eQTL associations between variants and genes using publicly available data57,58, and gene targets of distal enhancers using looping chromatin59.
Expression analysis
Gene expression and genotype data for 278 thyroid tissues from the GTEx Release phs000424.v6. p1 from 5.10.2015 (GTEx Analysis V6) were downloaded from the GTEx consortium57 via dbGAP. Linear regression analysis for expression of SMAD3 and genotypes of rs73227498 and rs821749 on 5q22.1 as well as for rs2289261 and rs56062135 on 15q22.33 was performed using quantile normalized expression values and the following covariates from GTEx: 3 genotyping principal components, gender, genotyping platform (Illumina’s OMNI 5M or 2.5M array) and 35 PEER factors.
Data availability
The Icelandic population WGS data have been deposited at the European Variant Archive under accession code PRJEB8636. The authors declare that the data supporting the findings of this study are available within the article, its Supplementary Information files and on request.
Additional information
How to cite this article: Gudmundsson, J. et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 8, 14517 doi: 10.1038/ncomms14517 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Howlader, N. et al. SEER Cancer Statistics Review, 1975-2013 National Cancer Institute http://seer.cancer.gov/csr/1975_2013/ based on November 2015 SEER data submission, posted to the SEER website, April (2016).
Engholm, G. et al. NORDCAN: cancer incidence, mortality, prevalence and survival in the nordic countries, Version 7.3 (08.07.2016). Association of the Nordic Cancer Registries. Danish Cancer Society. http://www.ancr.nu accessed on 20/12/2016.
Kondo, T., Ezzat, S. & Asa, S. L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 6, 292–306 (2006).
DeLellis, R. A. Pathology and genetics of thyroid carcinoma. J. Surg. Oncol. 94, 662–669 (2006).
Goldgar, D. E., Easton, D. F., Cannon-Albright, L. A. & Skolnick, M. H. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J. Natl Cancer Inst. 86, 1600–1608 (1994).
Czene, K., Lichtenstein, P. & Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int. J. Cancer 99, 260–266 (2002).
Amundadottir, L. T. et al. Cancer as a complex phenotype: pattern of cancer distribution within and beyond the nuclear family. PLoS Med. 1, e65 (2004).
Gudmundsson, J. et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 41, 460–464 (2009).
Gudmundsson, J. et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat. Genet. 44, 319–322 (2012).
Figlioli, G. et al. A comprehensive meta-analysis of case-control association studies to evaluate polymorphisms associated with the risk of differentiated thyroid carcinoma. Cancer Epidemiol. Biomarkers Prev. 25, 700–713 (2016).
Tcheandjieu, C. et al. Fine-mapping of two differentiated thyroid carcinoma susceptibility loci at 9q22.33 and 14q13.3 detects novel candidate functional SNPs in Europeans from metropolitan France and Melanesians from New Caledonia. Int. J. Cancer 139, 617–627 (2016).
Wang, Y. L. et al. Confirmation of papillary thyroid cancer susceptibility loci identified by genome-wide association studies of chromosomes 14q13, 9q22, 2q35 and 8p12 in a Chinese population. J. Med. Genet. 50, 689–695 (2013).
Landa, I. et al. The variant rs1867277 in FOXE1 gene confers thyroid cancer susceptibility through the recruitment of USF1/USF2 transcription factors. PLoS Genet. 5, e1000637 (2009).
Takahashi, M. et al. The FOXE1 locus is a major genetic determinant for radiation-related thyroid carcinoma in Chernobyl. Hum. Mol. Genet. 19, 2516–2523 (2010).
Sveinbjornsson, G. et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet. 48, 314–317 (2016).
Genome of the Netherlands, C. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 46, 818–825 (2014).
Ge, M. et al. Functional evaluation of TERT-CLPTM1L genetic variants associated with susceptibility of papillary thyroid carcinoma. Sci. Rep. 6, 26037 (2016).
Kohler, A. et al. Genome-wide association study on differentiated thyroid cancer. J. Clin. Endocrinol. Metab. 98, E1674–E1681 (2013).
Figlioli, G. et al. Novel genome-wide association study-based candidate loci for differentiated thyroid cancer risk. J. Clin. Endocrinol. Metab. 99, E2084–E2092 (2014).
Figlioli, G. et al. Novel genetic variants in differentiated thyroid cancer and assessment of the cumulative risk. Sci. Rep. 5, 8922 (2015).
Mancikova, V. et al. Thyroid cancer GWAS identifies 10q26.12 and 6q14.1 as novel susceptibility loci and reveals genetic heterogeneity among populations. Int. J. Cancer 137, 1870–1878 (2015).
Casteel, D. E. et al. A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J. Biol. Chem. 284, 5807–5818 (2009).
Miyake, Y. et al. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell 36, 193–206 (2009).
Levy, D. et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc. Natl Acad. Sci. USA 107, 9293–9298 (2010).
Codd, V. et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 45, 422–427 427e1-2 (2013).
Law, M. H. et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 47, 987–995 (2015).
Fingerlin, T. E. et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 45, 613–620 (2013).
Figueroa, J. D. et al. Genome-wide association study identifies multiple loci associated with bladder cancer risk. Hum. Mol. Genet. 23, 1387–1398 (2014).
Chubb, D. et al. Common variation at 3q26.2, 6p21.33, 17p11.2 and 22q13.1 influences multiple myeloma risk. Nat. Genet. 45, 1221–1225 (2013).
Swaminathan, B. et al. Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat. Commun. 6, 7213 (2015).
Codd, V. et al. Common variants near TERC are associated with mean telomere length. Nat. Genet. 42, 197–199 (2010).
Prescott, J. et al. Genome-wide association study of relative telomere length. PLoS ONE 6, e19635 (2011).
Luhrig, S. et al. Lrrc34, a novel nucleolar protein, interacts with npm1 and ncl and has an impact on pluripotent stem cells. Stem Cells Dev. 23, 2862–2874 (2014).
Bojesen, S. E. et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 45, 371–384 384e1-2 (2013).
Kote-Jarai, Z. et al. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum. Mol. Genet. 22, 2520–2528 (2013).
Jendrzejewski, J. et al. Papillary thyroid carcinoma: association between germline DNA variant markers and clinical parameters. Thyroid 26, 1276–1284 (2016).
He, H. et al. Multiple functional variants in long-range enhancer elements contribute to the risk of SNP rs965513 in thyroid cancer. Proc. Natl Acad. Sci. USA 112, 6128–6133 (2015).
He, H. et al. Genetic predisposition to papillary thyroid carcinoma: involvement of FOXE1, TSHR, and a novel lincRNA gene, PTCSC2. J. Clin. Endocrinol. Metab. 100, E164–E172 (2015).
Gulcher, J. R., Kristjansson, K., Gudbjartsson, H. & Stefansson, K. Protection of privacy by third-party encryption in genetic research in Iceland. Eur. J. Hum. Genet. 8, 739–742 (2000).
Galesloot, T. E. et al. Cohort Profile: the Nijmegen Biomedical Study (NBS). Int J Epidemiol doi:10.1093/ije/dyw268 (2016).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Kong, A. et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 40, 1068–1075 (2008).
Kong, A. et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103 (2010).
Delaneau, O., Howie, B., Cox, A. J., Zagury, J. F. & Marchini, J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 93, 687–696 (2013).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Kutyavin, I. V. et al. A novel endonuclease IV post-PCR genotyping system. Nucleic Acids Res. 34, e128 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913 (2007).
Mantel, N. & Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl Cancer Inst. 22, 719–748 (1959).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Consortium, E.P. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011).
Chadwick, L. H. The NIH Roadmap Epigenomics Program data resource. Epigenomics 4, 317–324 (2012).
Sheffield, N. C. et al. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 23, 777–788 (2013).
Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Whalen, S., Truty, R. M. & Pollard, K. S. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 48, 488–496 (2016).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010).
Acknowledgements
We thank the patients whose contribution made this work possible. The study was supported in part by grants from the National Institutes of Health: CA16058 (to the Ohio State University Comprehensive Cancer Center) and CA168505 (to M.D.R. and A.d.l.C.). This work was supported in part by The University of Texas MD Anderson Cancer Center start-up funds (to E.M.S.); an American Thyroid Association research grant (to E.M.S.), National Institutes of Health (NIH) grant U01 DE019765-01 (to E.M.S.); and Cancer Center Support Grant CA016672 to the University of Texas MD Anderson Cancer Center. This study makes use of data generated by the Genome of the Netherlands Project (GoNL). A full list of the investigators is available from http://www.nlgenome.nl. Funding for the GoNL project was provided by the Netherlands Organization for Scientific Research under award number 184021007, dated 9 July 2009 and made available as a Rainbow Project of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRI-NL). The sequencing was carried out in collaboration with the Beijing Institute for Genomics (BGI). This study also makes use of data generated by the GTEx Consortium and we are grateful for early stage sharing of RNA-seq and genotypic data. The following acknowledgement refers to the GTEx data: The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health. Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI\SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171) and Science Care, Inc. (X10S172). The Laboratory, Data Analysis and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to The Broad Institute, Inc. Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The Brain Bank was supported by supplements to University of Miami grants DA006227 and DA033684 and to contract N01MH000028. Statistical Methods development grants were made to the University of Geneva (MH090941 and MH101814), the University of Chicago (MH090951, MH090937, MH101820 and MH101825), the University of North Carolina—Chapel Hill (MH090936 and MH101819), Harvard University (MH090948), Stanford University (MH101782), Washington University St Louis (MH101810) and the University of Pennsylvania (MH101822).
Author information
Authors and Affiliations
Contributions
The study was designed and results were interpreted by J.G., G.T., U.T., T.R. and K.S. Statistical analysis was carried out by G.T., D.F.G., G.M., J.K.S., L.S., G.H.H., H. Helgason, P.S. and J.G. Subject recruitment, biological material collection and handling was organized and carried out by J.G., J.G.J., S.N.S., L.S., H. Helgason, S.L., M.D.R., E.A., A.P., E.P., A.G.-C., A.De.J., F.R., L.X., G.I.E., I.O., O.S., H.K. J.G., L.A.K., R.T.N.-M., T.S.P., T.J., H. Hjartarson, J.H., J.I.M., E.M.S., A.d.l.C., U.T. and T.R. Genotyping was supervised and carried out by J.G, H.J. and U.T. Analysis of expression data was done by J.G., G.H.H. and H. Helgason. Bioinformatics analysis was done by S.A.G. and J.G. Authors J.G., G.T. and K.S. drafted the manuscript. All authors contributed to the final version of the paper. Principal collaborators for the non-Icelandic study groups were J.I.M. (Spain), E.M.S. (Houston, USA), A.d.l.C. (Columbus, USA) and R.T.N.-M. and L.A.K. (the Netherlands).
Corresponding authors
Ethics declarations
Competing interests
The authors from deCODE genetics/AMGEN are employees of deCODE genetics/AMGEN. The other authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures, Supplementary Tables, and Supplementary References (PDF 2160 kb)
Supplementary Data 1
Results from meta-analysis for GWAS-identified risk variants by other groups (XLSX 16 kb)
Supplementary Data 2
Functional annotation of lead and correlated variants at GWAS risk loci (r2=0.75 in Icelandic population) from Haploreg, ENCODE, Ensembl, GtEX, TargetFinder, Blood eQTL browser and Regulatory Elements Database (XLSX 31 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gudmundsson, J., Thorleifsson, G., Sigurdsson, J. et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat Commun 8, 14517 (2017). https://doi.org/10.1038/ncomms14517
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms14517
This article is cited by
-
Polygenic risk scores, radiation treatment exposures and subsequent cancer risk in childhood cancer survivors
Nature Medicine (2024)
-
Multi-trait analysis characterizes the genetics of thyroid function and identifies causal associations with clinical implications
Nature Communications (2024)
-
CaMKK2 and CHK1 phosphorylate human STN1 in response to replication stress to protect stalled forks from aberrant resection
Nature Communications (2023)
-
Genetic susceptibility to hereditary non-medullary thyroid cancer
Hereditary Cancer in Clinical Practice (2022)
-
Implementation of individualised polygenic risk score analysis: a test case of a family of four
BMC Medical Genomics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
