
Abstract
Oceanic oxygen minimum zones are strong sources of the potent greenhouse gas N2O but its microbial source is unclear. We characterized an exponential response in N2O production to decreasing oxygen between 1 and 30 μmol O2 l−1 within and below the oxycline using 15NO2−, a relationship that held along a 550 km offshore transect in the North Pacific. Differences in the overall magnitude of N2O production were accounted for by archaeal functional gene abundance. A one-dimensional (1D) model, parameterized with our experimentally derived exponential terms, accurately reproduces N2O profiles in the top 350 m of water column and, together with a strong 45N2O signature indicated neither canonical nor nitrifier–denitrification production while statistical modelling supported production by archaea, possibly via hybrid N2O formation. Further, with just archaeal N2O production, we could balance high-resolution estimates of sea-to-air N2O exchange. Hence, a significant source of N2O, previously described as leakage from bacterial ammonium oxidation, is better described by low-oxygen archaeal production at the oxygen minimum zone’s margins.
Similar content being viewed by others


Introduction
Permanent oceanic oxygen minimum zones (OMZs) are significant sources of tropospheric N2O (∼0.8–1.35 Tg N yr−1 or 20–75% of oceanic total1 excluding coasts2), a potent greenhouse gas that also plays key roles in atmospheric chemistry1,3. Oversaturation of N2O within an OMZ is undoubtedly due to microbial activity but the precise nature of the organisms and biochemistry responsible for its production remain to be fully characterized. Some N2O production in OMZs has been ascribed to classic, canonical denitrification at the base of an oxycline4,5, whereas deeper into the functionally anoxic core of an OMZ, there is also net reduction of N2O to N2 by denitrification5,6,7. Above the anoxic core, production of N2O is traditionally described as a single function of bacterial nitrification under oxygen stress, with the yield of N2O, from the oxidation of ammonium, increasing as oxygen declines8,9,10. More recently, there have also been suggestions for a coupling (both inter- and intracellular) between denitrification and nitrification as a means of N2O production and there is growing evidence for a direct contribution from the Archaea to this process2,4,11,12,13.
The documented thickening of OMZs across the world14 has not only increased the volume of low oxygen waters with a potential to produce N2O but such thickening also makes that N2O more readily exchangeable with the atmosphere. There is clearly therefore a need to improve our understanding of the production of this atmospherically potent N2O at both the margins of OMZs and beyond in hypoxic, coastal waters2. Many have taken the linear negative correlation often observed between N2O (oversaturation relative to atmospheric equilibration) and O2 in surface waters to indicate bacterial nitrification as the predominant source of N2O (that is, the N2O anomaly versus apparent oxygen utilization and see ref. 15). Classic bacterial nitrification as the source of N2O was corroborated by early observations with a pure culture of the ammonia-oxidizing bacterium Nitrosomonas sp., where the yield of N2O per mole of ammonium oxidized increased exponentially as oxygen declined8. This regulatory effect of O2 on N2O production in the ocean is now widely accepted (some 100 papers citing8 in relation to ocean N2O production), though its exponential form has not, to the best of our knowledge, been characterized experimentally in the ocean below 30 μmol O2 l−1 (ref. 13). In addition, there are few, if any, ocean-based experimental data to substantiate this single physiological response. For example, incubation of OMZ oxycline waters with 15NH4+ might be expected to yield predominantly 46N2O, that is, both N in N2O derived from NH4+, (15NH4+→15NH2OH→15NO+15N2O→15NO2−), if classic oxygen-stressed, bacterial-nitrifier N2O production was active, but this is not the case4. Rather, this pathway of bacterial-nitrifier N2O production has routinely been used for the purposes of mass balance or to simply rationalize water column distributions of N2O without any supporting experimental evidence9,10.
In addition to the poorly substantiated mechanistic basis for bacterial-nitrifier N2O production in the ocean, it is now evident that the Archaea are widespread in the ocean, playing significant roles in key processes such as nitrification and potentially the production of N2O (refs 12, 13, 16, 17). The Thaumarchaeota, the archaeal phylum that encompasses the ammonium oxidizing archaea (AOA18), are commonly found in low-oxygen waters at the margins of an OMZ. AOA abundance decreases as oxygen concentrations rise towards air saturation in the upper mixed layers of the ocean13,19 and have been shown to also decrease as oxygen practically disappears at the oxic to anoxic interface at the core of an OMZ6. Thus, lower-oxygen waters appear to be an important niche for at least some Thaumarchaeota groups20. It is well established, at least for laboratory cultures, that bacterial ammonia oxidizers can produce N2O directly: either as a by-product of nitrification8 or through nitrifier–denitrification21. It is now evident that some AOA can also produce N2O during nitrification, but probably not via a nitrifier–denitrification-like process. New models of archaeal ammonia oxidation indicate a key role for NO produced by nitrite reductase encoded by the gene AnirK22 providing a clear link to the production of N2O, possibly via hybrid N2O formation22,23,24,25. A role for AOA-mediated N2O production has been suggested in the oceans, with process, natural abundance isotope values and molecular data supporting this idea12,20,26,27. There has, however, been no formal experimental characterization of N2O production at oxygen concentrations representative of the margins of an OMZ (<62.5 μmol O2 l−1) where N2O accumulates2,28 and/or the abundance of AOA (or any other candidate organisms) in representative samples of the ocean13.
Here we provide experimental evidence, with samples from the eastern tropical North Pacific (ETNP), which clearly links an exponential increase in N2O production to decreasing oxygen between 1 to 30 μmol l−1 and to archaeal gene abundance, together with a 15N pattern in the N2O that is reconciled by an archaeal mode of production.
Results
Water column characteristics
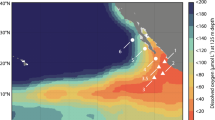
Along our offshore transect through the nitrite maximum zone (NMZ, Fig. 1a and Supplementary Table 1), the mixed layer depth (MLD) extended down to approximately 20 to 25 m and then the density increased steadily to a sharp inflection at 35 to 40 m, marking the base of the pycnocline (Fig. 1b). Oxygen dropped rapidly in the pycnocline to ≤50 μmol O2 l−1 at its base but remained above the limit of detection for the Seabird Sensor to approximately 340 m (Fig. 1c). Within these low-oxygen waters (6.8 μmol O2 l−1, median value) we measured a broad, characteristic peak in N2O of up to 104 nmol N2O l−1 (Fig. 1d). Deeper, at around 350 m, oxygen became comparatively constant, with the functionally anoxic core of the OMZ7, where both the secondary nitrite maxima and N2O minima were measured, occurring deeper still at 400 to 450 m (Supplementary Fig. 1a). We found that the waters at all of the depths described so far were supersaturated with CO2 (Supplementary Fig. 1b), with CO2 being strongly correlated with N2O over the top 45 m (Supplementary Fig. 1c).

(a) Offshore transect (bold black line with sites as filled red circles) through the nitrite maximum zone (NMZ) from 13 °N, at approximately 120 km off the coast, to 8 °N, at 670 km offshore. The red line marks the boundary of the permanent OMZ (min. O2<20 μmol l−1,) and the colour indicates the intensity of the nitrate deficit (>10 μmol l−1) within the NMZ (reprinted from ref. 80). (b) Density as a function of depth: upper light-blue line, approximate mixed layer depth (MLD); and lower light-blue line, base of the pycnocline. (c) Oxygen dropped rapidly in the pycnocline to ≤50 μmol O2 l−1 at its base (upper light-blue line) but remained above the limit of detection for the Seabird Sensor (∼ 1.6 μmol l−1) to 340 m (lower light-blue line). Within these boundaries (light-blue lines in c,d), oxygen was present at 6.8 μmol l−1, on average (median, vertical dark-blue), and we measured a broad peak in N2O (d). Profiles to 700 m are given in Supplementary Fig. 1. Panels b–d were drawn in SigmaPlot (Systat Software, San Jose, CA, USA).
We set our observations for N2O into the wider context of the eastern tropical North Pacific by comparing them with profiles in the MEMENTO database29 (Supplementary Fig. 2). Although there is considerable variation in the profiles, peak concentrations of 60 to 100 nmol N2O l−1 at 100 m are present between approximately 0 °N to 22 °N and out to approximately 155 °W, with this triangle roughly marking the extent of the nitrite maximum zone (NMZ), within the wider boundary of the OMZ as a whole30.
Nitrous oxide production as a function of oxygen
We measured the production of 15N2O in incubations with 15NO2− at two depths at each of our six offshore sites (n=12 groups of experiments). To generate natural variation in ambient water column oxygen and nitrous oxide concentrations, each depth was either within or beneath the oxycline (Supplementary Table 2). Each group of experiments comprised up to six oxygen treatments, giving us 70 independent observations (n=70) for the production of N2O as a function of oxygen (Table 1). Production of N2O (pN2Ototal equations 1, 2, 3, 4, 5) was strongly modulated by the level of oxygen in each treatment (likelihood ratio test for treatment, degree of freedom 5, χ2 38.365, P<0.0001 (ref. 7) and degree of freedom 3, χ2 34.688, P<0.0001 for the full and pooled data sets, respectively) and was maximal in waters degassed with nitrogen (Fig. 2a,b). In addition, the 15N2O produced in each experiment was predominantly single labelled 45N2O (that is, only having one 15N), at a level far above (81%, on average) that expected for denitrification (4–23%; Supplementary Table 1) given the 15N labelling of the NO2− pool (Fig. 2c, equations 2 and 3 in the ‘Methods’ section and Supplementary Table 1). Given the labelling of produced N2O, we could only ascribe 19% of the N2O to the reduction of exogenous nitrite (N2Oexogenous, equation 1) with the large majority (81%) of the N2O being due to some form of endogenous coupling (N2Oendogenous, equation 4 and see below).

(a) All oxygen treatments, including any inhibitory effect of N2O (50 nmol N2O l−1 crossed with O2). (b) As this latter treatment had no effect, the data were pooled by their comparative oxygen concentrations (3+4 and 5+6; see Table 1). Each box in a and b shows the 25th and 75th percentile, overall spread in the data and median value (horizontal line). In both a and b, the effect of treatment is highly significant (P<0.0001). (c) Production of p45N2O is clearly above that expected (p45N2Oexp) from denitrification of NO2− (see the ‘Methods’ section) in each treatment and the symbol colours in c are the same as in b (grey=100% air; red=ambient; green=OFN and N2O; olive-green=O2 and N2O+O2). Drawn in SigmaPlot (Systat Software, San Jose, CA, USA).
Ambient controls were used to represent N2O production in unadulterated water, that is, straight from the sampling bottles on the conductivity-temperature-depth rosette and, in these, oxygen concentrations ranged naturally from 1 to 199 μmol O2 l−1. In addition, the concentration of oxygen set at each level of the treatment varied (coefficient of variation (CV) of 4% to 35% for all the levels) across the 12 groups of experiments (Table 1). To account for this oxygen gradient in the 12 groups of experiments, and any natural variation in the water samples, we used a nonlinear mixed-effects approach to model the production of total N2O as an exponential function of decreasing oxygen (Table 2 and Supplementary Fig. 3). The most parsimonious model (M2) required only a random intercept (a), which allowed the overall magnitude of total N2O production to vary randomly between the 12 groups of experiments, while keeping the response to oxygen (b) constant. The mixed-effects model captures the data well (Fig. 3). The overall exponential increase in production of N2O with oxygen decreasing below 30 μmol O2 l−1 is not only consistent with N2O accumulating below 30 μmol O2 l−1 in the water column (Fig. 1d) but also with distributions seen in many parts of the tropical North Pacific (as above, Supplementary Fig. 2). We also measured the production of N2O over time at two oxygen concentrations (Supplementary Fig. 4) to check whether our 72 h incubation overestimated production. Where production was strongest (30 and 56 nmol m−3 d−1) and representative of the 12 main experiments (median 58 nmol m−3 d−1, <30 μmol O2 l−1), it was approximately linear over the first 18 h and then decreased over time. If anything, our single time point incubations may have underestimated N2O production slightly. Overall, however, we conclude that our experiment captured the regulation of N2O production by oxygen in the ocean. None of the incubations produced any 15N labelled N2, not even at 1 μmol O2 l−1.

Output from the nonlinear mixed-effects model M2 (Table 2) with (a) predicted production of N2O as a function of measured oxygen in each incubation bottle and (b) the same predicted production of N2O as in a, repeated as a function of measured production (95% confidence interval). Inset (c) the original data for the 12 ambient, unadulterated incubations (same units as a). Overall, by allowing any natural variation in the production of N2O (intercept a) to vary randomly between the 12 experiments, the nonlinear mixed-effects model captures the exponential increase in N2O production below 30 μM oxygen well. See main text for further explanation and Supplementary Fig. 3 for the complete model output with individual fits for each experiment and overall population parameter estimates. The latter of which we then use as input to a 1D model of water column N2O production. Drawn in SigmaPlot (Systat Software, San Jose, CA, USA).
Nitrification was also clearly active in the water column. We measured a primary nitrite maximum (Supplementary Fig. 1a) and, in ambient samples of water, with oxygen at 1 to 23 μmol O2 l−1, significant oxidation of both ammonium and nitrite (2.4 nmol N l−1 d−1 and 19.1 nmol l−1 d−1 on average for each, respectively), and net nitrification of up to 8.2 nmol N l−1 d−1 (Supplementary Table 2).
Variation in nitrous oxide production with gene abundance
By allowing the magnitude of total N2O production (a) to vary randomly between the 12 groups of experiments the mixed-effects model was able to derive an overall ‘population’ estimate for the response to decreasing oxygen. The magnitude of any deviation from this estimate, that is, the random intercept, can be used to further explore relationships with other explanatory variables that could potentially account for that random variation in the magnitude of N2O production. For example, there was a clear effect of ambient oxygen concentration in the water column: with samples collected from above 30 μmol O2 l−1 producing less N2O, on average, to those collected from below 30 μmol O2 l−1 (Supplementary Fig. 5a). In agreement with a growing number of cases in the literature, we were not able to detect either of the bacterial ammonia mono-oxygenase genes, β-amoA or γ-amoA, but we did find high abundance of archaeal amoA (AamoA, 5.6 × 103 copies ml−1, on average). We also quantified archaeal nirK (AnirK, 2.2 × 103 copies ml−1, on average) and bacterial nirK and nirS (13 × 103 and 0.7 × 103 copies ml−1, on average, respectively), genes coding for the potential to reduce NO2− (Supplementary Fig. 6). Both pairs of either archaeal or bacterial functional genes were positively correlated with each other but the pairs were ordinated separately in the samples collected (Supplementary Fig. 7). There was no visual indication of a pattern in the deviation of the random intercept and abundance of either bacterial nirK or nirS (Supplementary Fig. 5b) but there was a positive pattern in the abundance of AnirK and AamoA (Supplementary Fig. 5c) that we explore further.
The fully parameterized (oxygen combined with all four candidate genes) nonlinear mixed-effects models failed to converge and to explore the production of N2O as a function of both oxygen and functional gene abundance further, we log-transformed the data and proceeded with multiple linear regression (Supplementary Table 3). Oxygen alone had a highly significant negative effect on the production of N2O (Fig. 4a, Supplementary Table 3: M7 versus M6). Despite the comparatively similar abundance of bacterial nirS to the archaeal genes and the greater gene abundances for nirK, neither nirS nor nirK improved the fit of the model over oxygen alone, either singularly or when combined (Supplementary Table 3: M7 versus M8, M9, M10). Only inclusion of AamoA and/or Anirk in the model indicated any significant influence on the overall production of N2O (Supplementary Table 3: M7 versus M12, M13, M14, M15). As the model could not distinguish between the influence of either AamoA or Anirk on the distribution of the data, we would conclude that the most parsimonious explanation of our data is maximal production of N2O at lowest oxygen, combined with a positive influence from the abundance of both archaeal functional genes (Fig. 4). To confirm that the rate of N2O production was reasonable for the abundance of genes AnirK and AamoA detected, we calculated a per copy rate (equivalent to a per cell rate) for median N2O production (58 nmol m−3 d−1), below 30 μmol O2 l−1. Accordingly, 2 and 5 attomol N2O per copy per hour for the two genes, respectively, is representative of published rates (2–58 attomol N2O per cell per hour (ref. 31)).

(a) N2O production increasing with decreasing experimental oxygen (from Fig. 3a) and (b,c) production of N2O increasing as a function of the abundance of both AnirK and AamoA, that is, archaeal functional gene abundance accounts for some of the random variation in N2O production in the original nlme analysis (Table 2, Supplementary Fig. 5). The model (M14, Supplementary Table 3) suggested equal influence on the data from AnirK and AamoA, which the overwhelming 14N and 15N labelling of the N2O produced corroborated further. In a–c, the x axis data have been linearized and centred, see the ‘Methods’ section. Drawn in SigmaPlot (Systat Software, San Jose, CA, USA).
Depth-integrated N2O production and sea to air exchange
We used the coefficients from our nonlinear mixed-effects models (M2 and M5, Table 2) as input to a simple one-dimensional (1D) model of N2O, coded in R (ref. 32, see the ‘Methods’ section). The objective of this was to test whether a single O2-dependent, N2O production process could sustain the observed N2O maximum at∼100 m depth. Over a 30 day run (1 min time-step), parameterization with M2 maintained the initial steady state conditions (Fig. 5) without any marked accumulation (+0.04% d−1) of N2O, whereas, with M5, we saw significant N2O accumulation. Note that with M2 the vast majority of the N2O is assumed to come from a 1:1 coupling (equations 1, 2, 3, 4, 5), whereas with M5 we assume random mixing of 14NO and 15NO upstream of N2O production (equations 6 and 7). Given the better fit between our 1D model parameterized with M2, and our measured profiles of water column N2O, we favour and proceed with M2 (see the ‘Discussion’ section). The sea-surface N2O concentration in our model was fixed (Dirichlet boundary conditions) based on the average concentration from our observations (9.3 nmol N2O l−1). Sea-air exchange (efflux to the atmosphere) was therefore implicit in our model and this efflux of N2O was sustained by a positive concentration gradient with depth, implying an upward flux of N2O. Since the model water column was losing N2O at the surface, it is implicit that this upward flux of N2O should compensate for the gas exchange loss term (that is, equal sea-air exchange). Therefore, we calculated the flux over the upper 2 and 5 m of the model water column to derive the model sea-air flux. These depths were chosen to represent the turbulent layer near the sea surface given the relatively low average wind speed (5 m s−1, Supplementary Fig. 8b). The resulting sea-air flux of N2O was 17.9 μmol N2O m−2 d−1 and 16.0 μmol N2O m−2 d−1 over 2 and 5 m, respectively. We compare this with an estimate from our high-resolution pCO2 data (n=4,820) and water column profile data (‘Methods’ section and Supplementary Figs 1 and 8). Accordingly, our average estimate for CO2 exchange was 7.2 mmol CO2 m−2 d−1 (95% confidence interval of 6.9 to 7.4), with an equivalent exchange for N2O of 17 μmol N2O m−2 d−1 (95% confidence interval of 15.6 to 17.5); the latter agreeing well with the 1D model estimate for sea to air exchange of 16.0–17.9 μmol N2O m−2 d−1. Our experimental manipulation of oxygen, combined with a mixed-effects modelling approach has enabled us to parameterize a simple, single process 1D model that reproduces the pattern of N2O observed in the top 350 m of the tropical North Pacific.

(a) Concentration profiles for O2 and (b) for N2O. In b, N2O is either produced according to B15 from Babbin et al.5 or according to two parameterizations of equation (7), using either M2 or M5 (Table 2). In M2, we ascribe the majority of N2O production (pN2Ototal) to a 1:1 coupling, whereas, with M5, we assume that all of the N2O comes from a random mixing of 14N and 15N labelled NO (pN2Ototal′); note the better fit to the data with M2. (c) N2O production sources according to either a single response to oxygen, equation (7), parameterized with M2, or according to B15; where the two sources of nitrification and denitrification are B15 Nit and B15 Den, respectively. Initial conditions in both models were set using the mean profile from all of our observations but we show the output against all the data here to illustrate the goodness of fit of our simpler model. Drawn in SigmaPlot (Systat Software, San Jose, CA, USA).
Discussion
Here we have shown experimentally that production of N2O increases exponentially below 30 μmol O2 l−1 and that this sensitivity to oxygen holds along a 550 km offshore transect through the OMZ of the eastern tropical North Pacific. Further, variation in the overall magnitude of N2O production correlates positively with the abundance of archaeal functional genes that potentially play a role in that production of N2O. Parameterizing a simple 1D model with our experimentally derived exponential terms enabled us to model accurately the distribution of N2O over the top 350 m of the water column and, with a single response to oxygen, we could balance our estimates of sea to air exchange for N2O.
Here we were following up on our previous work in the Arabian Sea4 where the vast majority of N2O production could, apparently, be accounted for by canonical denitrification of 15N-NO2−. Accordingly, we enriched the NO2− pool with 15N in excess of 87 atom% and if denitrification were the dominant source of N2O, and the labelling of N2O random and binomially distributed (Supplementary Table 1), then we would have expected a maximum of 23% of the resulting N2O to be single-labelled, 45N2O. In contrast, however, we measured far more 45N2O than expected throughout (Fig. 2c). Put simply, the majority of N in the N2O produced was actually 14N that was not derived from our 15N-NO2− tracer. Although nitrite is known to have a stimulatory effect on the expression of nirK, at least in pure cultures of Nitrosomonas europaea, this is at nitrite concentrations of ∼10 mM, far higher than that applied here (10 μM), which is typical of 15N tracer work for 15N-gas production6,33,34.
One possibility is that dilution of the 15N-NO2− pool occurred via oxidation of 14N-NH4+ to 14N-NO2− and 15N-NO2− to 15N-NO3−, as this would increase the chance of any subsequent denitrification producing 45N2O, relative to 46N2O. For this to be plausible, however, 14N-NO2− production would need to make ∼20 μmol l−1 at the start of the incubation. We did measure significant nitrification activity (Supplementary Table 2) similar to that in the south eastern Pacific35. Such activity, however, could only turn over approximately 0.2% d−1 of the NO2− during our incubations, which would have had a negligible effect on the ratio of 45N2O to 46N2O produced. Alternatively, there could be a direct coupling between externally applied 15NO2− and internally supplied 14NO2−, or other 14N intermediate (for example, 14NO), from ammonia oxidation22,23, as shown for bacterial nitrifier–denitrification31. Such a coupling has been argued as a possible route for N2O production in the oligotrophic North Pacific36. Here we were not able to detect the bacterial ammonia mono-oxygenase genes, β-amoA or γ-amoA. Given this apparent absence of any bacterial, nitrifier–denitrifier genomic potential, along with the overestimation of N2O production in our model (M5, Fig. 5b), through such a path, we would refute bacterial nitrifier–denitrification in this setting. Finally, oxidation of NO2− has been measured simultaneously with NO3− reduction at up to ∼16 μmol O2 l−1 (ref. 35) with comparable activity of 14 nmol N l−1 d−1 and 21 nmol N l−1 d−1 for NO2− and NO3−, respectively (median values). Although we did not quantify NO3− reduction, if its activity were comparable to our measured rates of NO2− oxidation it would have the same negligible effect on the 15N labelling of the NO2− pool.
Our oxygen experiments, combined with statistical modelling, indicate that highest N2O production is best explained by low oxygen together with a high abundance of both AnirK and AamoA. Hence, the patterns in the genomic potential for both the reduction of NO2− (our source of 15N) and oxidation of ammonium (as a source of 14N, possibly NH2OH which has been reported as integral to archaeal ammonia oxidation22,23), combined with the concentration of O2, account for the predominant production of 14N and 15N labelled 45N2O. We could find no significant relationship for bacterial nirK and nirS. Admittedly, gene abundance (and by extrapolation cell number) does not necessarily confer a direct role on that gene for a measured process. Yet, similar positive relationships between Thaumarchaeota cell abundances and nitrification potentials are present in the low-oxygen waters of the Baltic, which, along with exponential increases in AamoA abundance below 100 μmol O2 l−1 in the Atlantic, suggest maintenance of active populations of Thaumarchaeota in low-oxygen waters13,19.
Overall, our data agree with the growing body of evidence for archaeal-mediated N2O production12,13. Although the precise biochemistry of this pathway is unknown, recent reports showing a key role for NH2OH and NO in Thaumarchaeotal ammonia oxidation provide support for a 1:1 coupling in N2O production in these organisms22,23 in support of our favoured model (Fig. 5b, M2, equations 1, 2, 3, 4, 5). As the nitrifying Archaea are better adapted to low oxygen compared with their bacterial analogues, it is unlikely N2O production occurs via an ammonia oxidizing bacterial type biochemical leak37. Rather, the metabolism of the potential precursor substrates (NH2OH and NO) in hybrid N2O formation might represent a genuine route of energy conservation22. Given the apparent absence of Nor or its equivalent in the archaea, we have to assume that our exogenous N2O (19% of total N2O production, on average) was produced through canonical denitrification operating as far as N2O but it remains to be proven whether the ability to metabolize NO further to N2O is truly absent from archaea in the ocean. Furthermore, modelling of N2O production in the ocean15 suggests that the oxycline is important, but has linked this N2O production primarily to bacterial ammonium oxidation (nitrification). Here we show that this is not the case and a major driver of N2O production in the ocean is likely to be archaeal hybrid N2O formation.
Although in principle, the regulatory effect of oxygen on N2O production in the ocean is widely accepted (some 100 papers citing Goreau et al.8 in relation to ocean N2O production), its exponential form has not, to the best of our knowledge, been characterized experimentally in the ocean below 30 μmol O2 l−1 (ref. 13). The basic linear ΔN2O/AOU relationship is frequently used to model the distribution of N2O across the oceans, where the slope represents the yield of N2O per mole of O2 consumed (see ref. 15 for a full discussion). This theoretical yield varies widely (0.076 to 0.31 nmol N2O μmol−1 O2 consumed) and can struggle to capture the full dynamics of N2O production in low-oxygen waters15. More fully parameterized versions of the ΔN2O/AOU that allow the yield to change as a function of oxygen do a better job but still struggle at the transition (notionally 4 μmol O2 l−1) from production to consumption of N2O (ref. 15). Here our mixed-effects modelling approach has enabled us to characterize a population estimate for the exponential increase in N2O production as a function of decreasing oxygen, from 1 μmol O2 l−1 to ∼30 μmol O2 l−1, without the confounding effects of individual site characteristics or, indeed, the need to invoke different metabolic pathways either side of an oxygen threshold.
The original version of the model by Babbin et al.5 required net production of N2O from both classic nitrification and denitrification to generate the typical 100 nmol l−1 peaks in N2O. Here, the orginal formulation of Babbin et al., however, generated net accumulation of 20–30% N2O (10–60 nmol l−1 over 30 days) below the pycnocline, whereas with our single process variant the model only gained 1.2%. This suggests that the dynamics of N2O production in the two studies were fundamentally different. Indeed, we did not measure any production of 15N2 in any of the oxygen treatments, that is, none of the 15N2O from the reduction of 15NO2− was further reduced to 15N2, even at 1 μmol O2 l−1, which is a key feature of the Babbin model. That, along with the non-binomial distribution of 15N in our N2O, relative to the NO2− pool, discounts denitrification as the primary source of N2O here and it is redundant in our model.
The two methods that we used to estimate sea to air exchange (17 μmol N2O m−2 d−1, on average from the N2O–CO2 field data and a 1D directly parameterized model) agreed very well with each other and with those in the literature—despite different approaches. For example, 13 μmol N2O m−2 d−1 has previously been taken as representative of the ETNP5, while a broader range of 5–31 μmol m−2 d−1 has been estimated for the tropical south Pacific2,38. What is important for estimating the contribution from the ocean to the global N2O budget is the respective area of OMZ used for any extrapolation. The latter is partly defined by the concentration chosen for oxygen at which the microbiology either produces or consumes N2O and this is contentious5,6. Here we have measured a clear exponential increase in N2O production with decreasing oxygen between 1 to 30 μmol O2 l−1 and apply this to regions of the ocean defined as OMZs by oxygen minima below 20 μmol O2 l−1 (ref. 30). Applying our average rate to the OMZ of the ETNP (12.4 × 1012 m2 including a coastal strip making up only ∼3.4% of the area) and the entire global extent of OMZs (30.4 × 1012 m2) generates 2.1 Tg N y−1 and 5.1 Tg N y−1, respectively, as N2O. The latter of which agrees very well with estimates of approximately 5.8 Tg N y−1 derived from the oxygen-sensitive model of Nevison et al.15
Modelling our single response of a predominantly archaeal-driven hybrid N2O formation process not only accurately reproduces the distribution of N2O over the top 350 m of the water column but this single response can also balance our high-resolution estimates of sea to air exchange for N2O. Hence, a significant source of N2O that has for a long time now been ascribed to bacterial-mediated ammonium oxidation leaking N2O under oxygen stress, can better be described by an archaeal-driven hybrid N2O formation process exploiting the niche of low oxygen waters, at the margins of an OMZ.
Methods
Site-specific water column profiles and underway pCO2 data
A standard conductivity–temperature–depth rosette (24 Niskin (20 litres) and full Sea-Bird 24 electronics (salinity, density, O2, temperature and so on) was used to collect and characterize the water at each site between 5 and 4,000 m. The distribution of N2O, CO2 and NO2− was measured as described previously4, except that the GC also had a hot-nickel catalyst and flame-ionization detector to quantify CO2 after rapid equilibration and reduction to CH4 (ref. 39). High temporal resolution measurements of pCO2 in surface seawater and atmosphere were also made every 5 min using an underway instrument (see below).
Production of N2O as a function of oxygen and gene abundance
We measured the production of 15N-N2O at two depths, both within and beneath the oxycline, at each of the six sites (Supplementary Table 1). Seawater was drained from a Niskin into 4 litre Nalgene bottles and sparged for 20 min to generate six oxygen treatments (Table 1). Seawater was then dispensed under pressure into 4 × 1 litre clear glass moulded infusion vials (Laboratory Precision Limited), except for the Ambient treatments, which went directly into the 1 litre vials. Oxygen (50 μm calibrated electrode, Unisense) and temperature were measured and, following up on studies in the Arabian Sea4, the vials spiked with 15N-NO2− ([10 μM], 98 atom%, Sigma, see Supplementary Table 1 for 15N atom % in each of the 12 sets of experiments). It is important to appreciate that all published work to date that uses 15N to trace the production of either N2O or N2 applies a ‘tracer’ at concentrations in excess of apparent Km values for these processes, that is, typically 5–10 μmol NO2− l−1 spike, compared with 1–2 μmol NO2− l−1 km and, as such, should be considered as potentials6,34,40. The vials were then sealed and incubated in the dark, at 12 °C, for 72 h as previously4. Later, bacterial activity was stopped in three of the vials by the addition of 6 ml formaldehyde (36% v/v), while the fourth vial was used to measure oxygen and the water then filtered (Supor, ø=47 mm, 0.2 μm pore size filters). The filters were immediately frozen in 2 ml cryovials, in liquid nitrogen, and stored at −80 °C for later extraction of nucleic acids (see below). Production of 15N2O and 15N2 was measured in the three remaining vials against reference samples for each of the treatments, or natural abundance, by mass spectrometry (see below and Nicholls et al.4). The data for each triplicate were then averaged and the mean value compared with its corresponding, single measure of functional gene abundances. Genes targeted with a potential role in N2O production were: β- and γ-proteobacterial amoA; bacterial nirS, bacterial nirK, archaeal nirK, archaeal amoA (here AamoA) and, in addition, general bacterial and archaeal Marine Group I 16S rRNA genes (see Supplementary Table 4 for primer sets). A combination of the large 1 litre glass vials and multiple oxygen treatments precluded a full time series incubation in each of the 12 N2O experiments (1,400 bottles versus 280). We did, however, measure N2O production at 2, 4, 9, 18, 36 and 72 h at two sites, for two oxygen treatments during a subsequent cruise to check that our single time point incubation was not overestimating production.
Mass spectrometry for 15N2O and 15N2 and rate calculations
All the samples were transferred under constant temperature back to the home laboratory in London and were brought to 22 °C before processing. Two subsamples of the 1 litre vials were forced out under helium and transferred to either a helium-filled 12 ml gas-tight vial (Exetainer, Labco), for 15N2 analysis, or a helium-filled 20 ml gas-tight vial (Gerstel and 20 mm butyl rubber stoppers and aluminium seals, Grace—Alltech) for 15N2O analysis. The 20 ml vials ended up with 10 ml of seawater and 10 ml of helium headspace to which we added a carrier of 3 nmol N2O, as sparging with the compressed air, N2 and O2 treatments effectively removed all of the natural N2O from the samples. These were then analysed for enrichment of both single- and dual-labelled 45N2O and 46N2O, respectively, against seawater samples (collected on the cruise) sparged with the five treatment gases, or, in the case of the ambient treatment, reference samples of seawater, using a trace gas pre-concentrator unit (PreCon, Thermo-Finnigan)4. Calibration was performed against known amounts of N2O (98 p.p.m.; BOC), and it was linear (r2=0.998) over the range 0 to 20.72 nmol N2O absolute (∑44N2O, 45N2O and 46N2O).
After bringing the remaining 12 ml gas-tight vials to 22 °C, a helium headspace (1 ml) was added and the vials shaken by hand and left overnight on rollers (Spiramix) to allow N2 gas to equilibrate between the water phase and headspace. Samples of the headspace (100 μl) were then analysed for enrichment in 15N2 by injection (Multipurpose Sampler MSP2, Gerstel) into an elemental analyzer (Flash EA 1112, Thermo-Finnigan), interfaced with the continuous flow isotope ratio mass spectrometer (CF-IRMS)4. Calibration was performed at the beginning of each run with known amounts of oxygen free nitrogen gas (BOC) in seawater collected on the cruise, in the range of 0 to 12.6 μmol N2 absolute (∑28N2, 29N2 and 30N2). Values for the production of 29N2 and/or 30N2 were calculated as excess over the production in the time zero ‘reference’ samples41.
We used 15NO2− to trace the production of N2O as per our previous work in the Arabian Sea4 and present two principle methods for calculating the total production of N2O in response to oxygen. In the first method, given that the archaea appear to lack Nor, we assume that they cannot make N2O purely from exogenous NO2− and that any measured production of p46N2O (that is, 2 × 15NO2−) must be due to canonical denitrification reducing NO2− as far as N2O. Then, any production of N2O that we cannot account for by canonical denitrification with exogenous NO2− we assign to hybrid N2O formation, as in the most recent models for Thaumarchaeotal ammonia oxidation22. In the second method, we assume that all of the measured production of N2O is due to a classic bacterial-type mode of nitrifier–denitrification, with random isotope pairing of 14NO and 15NO upstream of the production of N2O.
We calculate the overall production of N2O that we assume to be owing to canonical denitrification of exogenous NO2− according to:

where FNNO2− is the fraction of 15N in the NO2− pool (Supplementary Table 1) in each set of incubations, determined by difference34, and we ignore any turnover by either ammonium or nitrite oxidation, which is shown to be negligible relative to the size of the NO2− pool (See the ‘Discussion’ section and Supplementary Table 2). We then used the measured amount of dual-labelled p46N2O to predict the expected amount of single-labelled p45N2Oexp for canonical denitrification according to40:

We would then argue that any production of p45N2O above p45N2Oexp cannot be solely due to reduction of external NO2−, and must be due to 15N pairing with an alternative source of 14N (for example, 15NO from 15NO2−, pairing with 14NH2OH in archaeal hybrid N2O formation22) which, for simplicity, we refer to as endogenous N2O:


The first estimate of total production of N2O in our incubations with 15NO2− is then the sum of the two former products:

Hence, the calculation of pN2Ototal, pN2Oendogenous and pN2Oexogenous with 15NO2− is synonymous to that for total N2, anammox and denitrification, respectively, in all other work measuring the production of N2; though the biological context is not40. The alternative formulation assumes that all of our measured production of N2O was dominated by a classic bacterial-type mode of nitrifier–denitrification, with random isotope pairing of 14NO and 15NO upstream of the production of N2O (ref. 22) and we can calculate an alternative pN2Ototal′ according to42:


Molecular analysis
In the home laboratory, each Supor filter was cut in half and one half was placed into a 2 ml sterile screw-cap tube, containing ø=0.1 mm glass beads. The following solutions were then added to each tube: 700 μl of 120 mmol l−1 sodium phosphate (pH 8.0) plus 1% (w/v) acid-washed polyvinylpolypyrrolidone, 500 μl of Tris-equilibrated phenol (pH 8.0), and 50 μl of 20% (w/v) sodium dodecyl sulfate. The extraction process involved bead beating and passing the samples through hydroxyapatite and Sephadex G-75 spin columns, to separate nucleic acids from proteins and salts43. Nucleic acids were resuspended in 50 μl of TE (10 mmol l−1 Tris, 1 mM EDTA [pH 8.0]) and stored at −80 °C.
The extracted DNA was used for quantification of functional genes (primer details are shown in Supplementary Table 4). Quantitative real-time PCR was performed in a Bio-Rad CFX96 Real-Time System. The reaction was performed in duplicate in a final volume of 15 μl, which contained 7.5 μl of SensiFAST SYBR No-ROX mix (2 × ) (Bioline), 200 nmol l−1 of each primer and 1 μl of 10 times diluted DNA. The conditions for all reactions were as follows: 95 °C for 3 min; 40 cycles of 95 °C for 0.05 min and 60 °C for 0.30 min; 95 °C for 0.05 min; 65 °C for 0.05 min and a final step of 95 °C for 0.5 min. Absolute quantification of the targeted genes was performed with a series of 10-fold standard dilutions, using the CFX Manager version 2.0 software (Bio-Rad). Standards for bacterial 16S rRNA, nirS and nosZ genes were derived from Pseudomonas brenneri DSM15294; environmental PCR products were used for bacterial amoA, AamoA, nirK, AnirK and MG1 16S. Samples with Cq values that were the same or greater than those of the no template controls were assumed to be below the limit of detection (LOD). In each of these cases, the calculated LOD for the particular qPCR plate was used as the value for that sample (maximum LOD=171 copies ml−1). Specificity of the AnirK PCR was assessed by sequencing product from a number of sites. All showed that the PCR assay was specific for its target gene (data not shown).
Nitrification
To account for any possible turnover of the 15NO2− pool in our 72 h 15N2O incubations, we incubated additional water under ambient oxygen (1 to 23 μmol O2 l−1) from the second depth at each site (Supplementary Table 1). Water was sampled into 1 litre vials, allowed to overflow three times, sealed, brought to 12 °C and then, without any sparging, pushed out (2 mm Teflon tubing) under helium into the bottom of 12 ml, gas-tight vials (Exetainer, Labco), overflowed three times and sealed. The vials were then enriched from concentrated stocks (Sigma, sparged with OFN) to [10 μmol l−1], in quadruplets, with either 15NO2− or 15NH4+. Ammonia oxidation was estimated from the net accumulation of 15NO2− after the addition of 15NH4+, single time point incubations (96 h); nitrite oxidation from net accumulation of 15NO3− over 3, 6, 12, 24, 48 or 96 h from 15NO2− and overall net nitrification from the accumulation of total 15NOx− after 96 h from 15NH4+. The samples were fixed (50 μl 50% (w/v) ZnCl2) and production of 15NOx−, 15NO2− or 15NOx− measured with a sulphamic acid assay at the University of Southern Denmark.
Modelling N2O production
We formulated a simple 1D model of N2O (1 m depth resolution), coded in R. The model is largely based on the parameterizations given by Babbin et al.5 encompassing physical processes (upwelling, vertical diffusion and implicit gas exchange of N2O) as well as biological production of N2O. Vertical transport was parameterized according to Fickian diffusion with a diffusivity Kz of 4 × 10−4 m2 s−1 at the surface, decreasing linearly to 4 × 10−5 m2 s−1 at 10 m and remained constant thereafter apart from the pycnocline (20–48 m depth) where Kz was 1 × 10−5 m2 s−1. This Kz profile effectively simulated near surface turbulence while the remaining water column was dominated by diffusive processes. An upwelling velocity (wup) of 8 × 10−7 m s−1 and a particle sinking velocity (of 1.2 × 10−4 m s−1 (ref. 5) were used.
The model resolved the upper 400 m of the water column at 1 m depth resolution and 1 min time intervals. Boundary conditions at the surface and at 400 m were fixed and prescribed by the respective averages from our profiles. This average profile also described initial conditions for NO3−, PO43−, N2O and O2. Particulate Organic Carbon (POC) values for the ETNP were taken from the literature44,45, with a surface concentration of 3 μmol l−1, a sub-surface maximum of 5 μmol l−1 at 32 m and decreasing thereafter to 1.3 μmol l−1 at 400 m. Model POC remineralization (POCrem) was parameterized as a first-order process with a rate constant of 5 × 10−7 s−1. POC production at the surface was implicit via the fixed boundary concentration as in Babbin et al.5 In addition, we parameterized POC production (pPOCZ) at depth (Z) as a function of the upwelling NO3− flux and light attenuation:

where F is the ratio of upwelled NO3− used by primary producers (0.2), [NO3−] is the concentration of NO3− at depth Z, Kd is the light attenuation coefficient (0.09 m−1) and rN:Cremin is the N:C ratio production/remineralization (rN:Cremin=16/106). The value of Kd was chosen as it gave a subsurface pPOCZ maximum which was consistent with the positions of the subsurface POC- and chlorophyll-concentration maxima at the base of the mixed layer as observed during our cruise. NO3−, PO43− and O2 were linked to POC production/remineralization according to Redfield stoichiometry, as in Babbin et al.5 O2 consumption followed a respiratory ratio of ∼1.4 (rO:Crem=150:106).
Production to consumption of N2O was parameterized for two separate variants of the model: (i) according to Babbin et al.5 and (ii) as a function of O2 concentration as described here. All processes except those producing N2O were identical in both the models. In our second variant, we parameterized model N2O production (pN2O in nmol m−3 d−1) using the estimates for a and b from our nonlinear mixed-effects models (M2 and M5, Table 2 and equation 12):

Note that the original Babbin formulation included an [O2]-dependent Heaviside function which terminated N2O production when [O2]<0.4 μmol L−1. Here, as oxygen was always above 0.4 μmol O2 L−1 it was redundant and not included in our variant of the model.
Estimating N2O exchange using high-resolution pCO2 data
High temporal resolution measurements of pCO2 in surface seawater and atmosphere were made every 5 min using an underway instrument (PML Dartcom Live pCO2. UK.46,47) with the ‘vented’ equilibrator modification46. The equilibrator was fitted with two platinum resistance thermometers (Pico Technology, model PT100) and a water-jacket supplied with seawater from the ship’s underway seawater system. A seawater flow of 1.6 litres min−1 was maintained through the main equilibrator. The average warming between the ship’s underway seawater intake and the equilibrator was 0.2±0.1 °C. Atmospheric measurements of CO2 were taken from an intake located on the foremast. Both gas streams from the equilibrator headspace and the air inlet were dried in a Peltier cooler (−20 °C). Mixing ratios of CO2 and water in the marine air and equilibrator headspace were determined by non-dispersive infrared dection (LI-840, LI-COR). Measurements were referenced against secondary calibration gases (BOC Gases, UK) with known CO2 mixing ratios (257.6, 373.4 and 463.5 μmol CO2 per mole) in synthetic air mixtures (21% oxygen and 79% nitrogen). All calibration gases were calibrated against certified primary standards from the National Oceanic and Atmospheric Administration (244.9 and 444.4 μmol CO2 per mole). The pCO2 system described here showed high consistency with a similar pCO2 system and pCO2 calculated from independent TA, DIC and pH during ‘at sea’ inter-comparison48. Sampling was carried out continuously (every 5 min), with the exception of periods for maintenance. See Supplementary Fig. 7 for a summary of the pCO2 and wind data and resultant efflux estimates.
Then, for the samples for which high-resolution seawater pCO2 (pCO2sw) data were available but in which N2O was not directly quantified, we predicted molar seawater N2O concentrations (N2Osw) using the linear relationship between N2Osw and the molar seawater concentration of CO2 (CO2sw; Supplementary Fig. 1). To do this, we first estimated the CO2 and N2O solubility for each sample (mol kg−1 atm−1; refs 49, 50). CO2sw for each sample was next calculated as the product of its pCO2sw and corresponding molar solubility. Each resulting molar N2Osw concentration was then converted to pN2Osw by dividing it by the calculated, corresponding N2O molar solubility. Atmospheric pN2Oatm was taken as the average for samples collected from the bow of the ship throughout the ∼6 week cruise (348±6 natm s.e., n=35) and the corresponding N2O flux estimated from the high-resolution CO2 fluxes calculated using the average 12 h wind speed:


Statistical analyses
All the analyses were conducted in R (ref. 32) following procedures largely described in ref. 51. We began with linear mixed-effects models treating oxygen as a categorical variable and modelling N2O production as an additive, linear function of the six oxygen treatments (Table 1). With the linear mixed-effects models, we fitted the oxygen treatment as a fixed effect and included random intercepts for each of the 12 experiments, comparing models with and without ‘oxygen’ with likelihood ratio testing. Given that there was clear spread within the oxygen data, we then used nonlinear mixed-effects models to model 15N2O production as a continual, exponential function of oxygen:

Where pN2Ototal comes from equations (1, 2, 3, 4, 5) and O2exp is the measured concentration of oxygen (μmol l−1) in each incubation bottle and total production of N2O is that measured in each bottle at the end of its incubation. For the 12 sets of experiments analysed using nonlinear mixed-effects models, we either fitted both the intercept (a, that is, maximum N2O production) and sensitivity (b, that is, response to oxygen) as random effects, or, a and b, each individually, and compared model fit in each case with the Akaike Information Criterion (AIC). Relationships between these ‘random’ elements, that is, variance not explained by experimental oxygen and other possible explanatory variables (for example, gene abundance) were explored visually (at the 12-experiment, group level, n=12) and then more rigorously using multiple regression and the entire, linearized and centred (xc) data set (natural log, xc=x−xmean, n=70). Here, we judge the simplest model (that is, just oxygen) against more complex models (oxygen plus single or multiple functional gene abundance) also using likelihood ratio testing.
Data availability
The data that support the findings of this study are available from the authors on reasonable request, see author contributions for specific data sets.
Additional information
How to cite this article: Trimmer, M. et al. Nitrous oxide as a function of oxygen and archaeal gene abundance in the North Pacific. Nat. Commun. 7, 13451 doi: 10.1038/ncomms13451 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Bakker, D. E. et al. in Ocean-Atmosphere Interactions of Gases and Particles Springer Earth System Sciences eds Liss P. S., Johnson M. T. Ch. 3, 113–169Springer (2014).
Arevalo-Martinez, D. L., Kock, A., Loscher, C. R., Schmitz, R. A. & Bange, H. W. Massive nitrous oxide emissions from the tropical South Pacific Ocean. Nat. Geosci. 8, 530–533 (2015).
Dore, J. E., Popp, B. N., Karl, D. M. & Sansone, F. J. A large source of atmospheric nitrous oxide from subtropical North Pacific surface waters. Nature 396, 63–66 (1998).
Nicholls, J. C., Davies, C. A. & Trimmer, M. High-resolution profiles and nitrogen isotope tracing pairs reveal a dominant source of nitrous oxide and multiple pathways of nitrogen gas formation in the central Arabian Sea. Limnol. Oceanogr. 52, 156–168 (2007).
Babbin, A. R., Bianchi, D., Jayakumar, A. & Ward, B. B. Rapid nitrous oxide cycling in the suboxic ocean. Science 348, 1127–1129 (2015).
Dalsgaard, T. et al. Oxygen at nanomolar levels reversibly suppresses process rates and gene expression in Anammox and denitrification in the oxygen minimum zone off Northern Chile. Mbio 5, 1–14 (2014).
Thamdrup, B., Dalsgaard, T. & Revsbech, N. P. Widespread functional anoxia in the oxygen minimum zone of the Eastern South Pacific. Deep Sea Res. I 65, 36–45 (2012).
Goreau, T. J. et al. Production of NO2− and N2O by nitrifying bacteria at reduced concentrations of oxygen. Appl. Environ. Microbiol. 40, 526–532 (1980).
Dore, J. E. & Karl, D. M. Nitrification in the euphotic zone as a source for nitrite, nitrate, and nitrous oxide at Station ALOHA. Limnol. Oceanogr. 41, 1619–1628 (1996).
Codispoti, L. A. & Christensen, J. P. Nitrification, denitrification and nitrous-oxide cycling in the eastern tropical south-Pacific Ocean. Mar. Chem. 16, 277–300 (1985).
Freing, A., Wallace, D. W. R. & Bange, H. W. Global oceanic production of nitrous oxide. Philos. Trans. R. Soc. B Biol. Sci 367, 1245–1255 (2012).
Santoro, A. E., Buchwald, C., McIlvin, M. R. & Casciotti, K. L. Isotopic signature of N2O produced by marine ammonia-oxidizing Archaea. Science 333, 1282–1285 (2011).
Löscher, C. R. et al. Production of oceanic nitrous oxide by ammonia-oxidizing archaea. Biogeoscience 9, 2419–2429 (2012).
Stramma, L., Johnson, G. C., Sprintall, J. & Mohrholz, V. Expanding oxygen-minimum zones in the tropical oceans. Science 320, 655–658 (2008).
Nevison, C., Butler, J. H. & Elkins, J. W. Global distribution of N2O and the Delta N2O-AOU yield in the subsurface ocean. Glob. Biogeochem. Cycles 17, 1–18 (2003).
Lam, P. et al. Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc. Natl Acad. Sci. USA 104, 7104–7109 (2007).
Francis, C. A., Roberts, K. J., Beman, J. M., Santoro, A. E. & Oakley, B. B. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc. Natl Acad. Sci. USA 102, 14683–14688 (2005).
Brochier-Armanet, C., Boussau, B., Gribaldo, S. & Forterre, P. Mesophilic crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 6, 245–252 (2008).
Berg, C., Vandieken, V., Thamdrup, B. & Juergens, K. Significance of archaeal nitrification in hypoxic waters of the Baltic Sea. ISME J. 9, 1319–1332 (2015).
Lund, M. B., Smith, J. M. & Francis, C. A. Diversity, abundance and expression of nitrite reductase (nirK)-like genes in marine thaumarchaea. ISME J. 6, 1966–1977 (2012).
Wrage, N., Velthof, G. L., van Beusichem, M. L. & Oenema, O. Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol. Biochem. 33, 1723–1732 (2001).
Kozlowski, J. A., Stieglmeier, M., Schleper, C., Klotz, M. G. & Stein, L. Y. Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 10, 1836–1845 (2016).
Martens-Habbena, W. et al. The production of nitric oxide by marine ammonia-oxidizing archaea and inhibition of archaeal ammonia oxidation by a nitric oxide scavenger. Environ. Microbiol. 17, 2261–2274 (2015).
Zhalnina, K. V. et al. Genome sequence of Candidatus Nitrososphaera evergladensis from Group I.1b enriched from everglades soil reveals novel genomic features of the ammonia-oxidizing archaea. PLoS ONE 9, e101648 (2014).
Mosier, A. C., Lund, M. B. & Francis, C. A. Ecophysiology of an ammonia-oxidizing archaeon adapted to low-salinity habitats. Microb. Ecol. 64, 955–963 (2012).
Santoro, A. E., Casciotti, K. L. & Francis, C. A. Activity, abundance and diversity of nitrifying archaea and bacteria in the central California Current. Environ. Microbiol. 12, 1989–2006 (2010).
Wilson, S. T., del Valle, D. A., Segura-Noguera, M. & Karl, D. M. A role for nitrite in the production of nitrous oxide in the lower euphotic zone of the oligotrophic North Pacific Ocean. Deep Sea Res. I 85, 47–55 (2014).
Naqvi, S. W. A. et al. Marine hypoxia/anoxia as a source of CH4 and N2O. Biogeoscience 7, 2159–2190 (2010).
Bange, H. W. et al. MEMENTO: a proposal to develop a database of marine nitrous oxide and methane measurements. Environ. Chem. 6, 195–197 (2009).
Paulmier, A. & Ruiz-Pino, D. Oxygen minimum zones (OMZs) in the modern ocean. Prog. Oceanogr. 80, 113–128 (2009).
Shaw, L. J. et al. Nitrosospira spp. can produce nitrous oxide via a nitrifier denitrification pathway. Environ. Microbiol. 8, 214–222 (2006).
R. A language and environment for statistical computing R Foundation for Statistical Computing (2014).
Beaumont, H. J. E., Lens, S. I., Reijnders, W. N. M., Westerhoff, H. V. & van Spanning, R. J. M. Expression of nitrite reductase in Nitrosomonas europaea involves NsrR, a novel nitrite-sensitive transcription repressor. Mol. Microbiol. 54, 148–158 (2004).
Thamdrup, B. et al. Anaerobic ammonium oxidation in the oxygen-deficient waters off northern Chile. Limnol. Oceanogr. 51, 2145–2156 (2006).
Kalvelage, T. et al. Nitrogen cycling driven by organic matter export in the South Pacific oxygen minimum zone. Nat. Geosci. 6, 228–234 (2013).
Wilson, S. T., del Valle, D. A., Segura-Noguera, M. & Karl, D. M. A role for nitrite in the production of nitrous oxide in the lower euphotic zone of the oligotrophic North Pacific Ocean. Deep Sea Res. I 85, 47–55 (2014).
Jason, J., Cantera, L. & Stein, L. Y. Role of nitrite reductase in the ammonia-oxidizing pathway of Nitrosomonas europaea. Arch. Microbiol. 188, 349–354 (2007).
Farias, L. et al. Denitrification and nitrous oxide cycling within the upper oxycline of the eastern tropical South Pacific oxygen minimum zone. Limnol. Oceanogr. 54, 132–144 (2009).
Sanders, I. A. et al. Emission of methane from chalk streams has potential implications for agricultural practices. Freshwat. Biol 52, 1176–1186 (2007).
Thamdrup, B. & Dalsgaard, T. Production of N2 through anaerobic ammonium oxidation coupled to nitrate reduction in marine sediments. Appl. Environ. Microbiol. 68, 1312–1318 (2002).
Thamdrup, B. & Dalsgaard, T. The fate of ammonium in anoxic manganese oxide-rich marine sediment. Geochim. Cosmochim. Acta 64, 4157–4164 (2000).
Nielsen, L. P. Denitrification in sediment determined from nitrogen isotope pairing. FEMS Microbiol. Ecol. 86, 357–362 (1992).
Purdy, K. J., Embley, T. M., Takii, S. & Nedwell, D. B. Rapid extraction of DNA and rRNA from sediments by a novel hydroxyapatite spin-column method. Appl. Environ. Microbiol. 62, 3905–3907 (1996).
Martiny, A. C., Vrugt, J. A. & Lomas, M. W. Concentrations and ratios of particulate organic carbon, nitrogen, and phosphorus in the global ocean. Sci. Data 1, 140048–140048 (2014).
Wakeham, S. G. & Canuel, E. A. Organic geochemistry of particulate matter in the eastern tropical north Pacific Ocean—implications for particle dynamics. J. Mar. Res. 46, 183–213 (1988).
Kitidis, V. et al. Seasonal dynamics of the carbonate system in the Western English Channel. Cont. Shelf Res. 42, 30–40 (2012).
Hardman-Mountford, N. J. et al. An operational monitoring system to provide indicators of CO2 related variables in the ocean. ICES J. Mar. Sci. 65, 1498–1503 (2008).
Ribas-Ribas, M. et al. Intercomparison of carbonate chemistry measurements on a cruise in northwestern European shelf seas. Biogeoscience 11, 4339–4355 (2014).
Weiss, R. F. Carbon dioxide in water and seawater: the solubility of a non-ideal gas. Mar. Chem. 2, 203–215 (1974).
Weiss, R. F. & Price, B. A. Nitrous oxide solubility in water and seawater. Mar. Chem. 8, 347–359 (1980).
Pinheiro, J. C. & Bates, D. M. Mixed-Effects Models in S and S-Plus Springer (2000).
Acknowledgements
We thank I.A. Sanders for technical assistance and R. Nicholls and G. Yvon-Durocher for statistical guidance; the captain and crew of RRS Discovery for their support throughout the cruise (D373) and the Natural Environment Research Council (NERC) (NE/E01559X/1).
Author information
Authors and Affiliations
Contributions
M.T. and K.J.P. conceived the idea for the study, M.T. analysed the data and wrote the paper. P.-M.C. did all the cruise logistics/preparations and performed PreCon and IRMS. P.-M.C. and K.J.P. performed the molecular analyses. S.T.M. performed GC analysis on-board. V.K. remotely managed the pCO2 system and with M.T. parameterized the 1D model and R.C.U.-G. scaled the N2O using the pCO2. All the authors commented on and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8, Supplementary Tables 1-4 and Supplementary References (PDF 1002 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Trimmer, M., Chronopoulou, PM., Maanoja, S. et al. Nitrous oxide as a function of oxygen and archaeal gene abundance in the North Pacific. Nat Commun 7, 13451 (2016). https://doi.org/10.1038/ncomms13451
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms13451
This article is cited by
-
Structure and function of the soil microbiome underlying N2O emissions from global wetlands
Nature Communications (2022)
-
The contribution of nirK gene-containing thaumarchaea to denitrification and N2O production across coastal sediment and terrestrial ecosystems
Journal of Soils and Sediments (2022)
-
Microbial N2O consumption in and above marine N2O production hotspots
The ISME Journal (2021)
-
Cyanobacteria and cyanophage contributions to carbon and nitrogen cycling in an oligotrophic oxygen-deficient zone
The ISME Journal (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
