
Abstract
The identification of cancer-associated mutations in the tricarboxylic acid (TCA) cycle enzymes isocitrate dehydrogenases 1 and 2 (IDH1/2) highlights the prevailing notion that aberrant metabolic function can contribute to carcinogenesis. IDH1/2 normally catalyse the oxidative decarboxylation of isocitrate into α-ketoglutarate (αKG). In gliomas and acute myeloid leukaemias, IDH1/2 mutations confer gain-of-function leading to production of the oncometabolite R-2-hydroxyglutarate (2HG) from αKG. Here we show that generation of 2HG by mutated IDH1/2 leads to the activation of mTOR by inhibiting KDM4A, an αKG-dependent enzyme of the Jumonji family of lysine demethylases. Furthermore, KDM4A associates with the DEP domain-containing mTOR-interacting protein (DEPTOR), a negative regulator of mTORC1/2. Depletion of KDM4A decreases DEPTOR protein stability. Our results provide an additional molecular mechanism for the oncogenic activity of mutant IDH1/2 by revealing an unprecedented link between TCA cycle defects and positive modulation of mTOR function downstream of the canonical PI3K/AKT/TSC1-2 pathway.
Similar content being viewed by others


Introduction
It is now evident that aberration of normal metabolic function represents a hallmark of tumorigenesis1. For example, mutation, deletion or silencing of genes encoding tricarboxylic acid cycle (TCA cycle) proteins, including isocitrate dehydrogenases 1 and 2 (IDH1/2), are frequent in acute myeloid leukaemias and brain cancers (WHO grade 2 and 3 astrocytomas, oligodendrogliomas and secondary glioblastomas)2,3. IDH1/2 mutations occur predominantly at arginine 132 for IDH1, and arginine 140 or 172 for IDH2 (refs 2, 3), in each case resulting in a gain of function that promotes conversion of αKG to 2HG (refs 4, 5). These latter metabolites share high structural similarity, allowing 2HG to bind and inhibit αKG-dependent enzyme activity including that of the Jumonji lysine demethylases family and the Ten-eleven translocation (TET) family of 5-methylcytosine hydroxylases6,7.
Jumonji family members require αKG as co-substrate to catalyse the removal of methyl moieties on lysine. Conversely, 2HG has been shown to effectively inhibit this family of enzymes6,7. Jumonji proteins are oxygenases catalysing demethylation of mono-, di- or tri-methylated lysines8,9. To date, more than 30 Jumonji proteins have been identified, which carry different secondary domains in addition to their common JmjC catalytic domain. These enzymes are classified into numerous sub-families according to sequence homology and substrate specificity. The KDM4 sub-family comprises four members (KDM4A-D) which are Fe(II)- and α-ketoglutarate-dependent enzymes possessing a JmjC catalytic domain8,9. KDM4A-C display similar structures including JmjN-, tandem PHD-, and tandem Tudor-domains and each catalyses demethylation of di- and tri-methylated H3K9 and H3K36. On the other hand, KDM4D lacks the PHD and tandem Tudor domains and is specific for H3K9(me2/3) (refs 10, 11). KDM4A is involved in the regulation of many cellular processes including cell cycle progression, DNA damage signalling and Ras-induced senescence12,13,14.
The mammalian (mechanistic) Target Of Rapamycin (mTOR) is a serine/threonine kinase belonging to the phosphoinositide 3-kinase (PI3K)-related kinase (PIKK) family. mTOR associates with either Raptor or Rictor to form distinct complexes, that is, mTORC1 and mTORC2, respectively, both of which are regulated by growth factors, amino acids and energy levels15,16,17,18. PRAS40 is recovered within the mTORC1 complex, while mSin1 and Protor1/2 are present in the mTORC2 complex19,20,21,22. mLST8 and DEPTOR, an inhibitor of mTOR, are common to both mTORC1 and mTORC2 (refs 23, 24). Among the cellular processes regulated by mTORC1 are cell growth and autophagy, while mTORC2 regulates cell survival and cytoskeleton organisation (for a review, see ref. 25). mTORC1/2 are frequently activated in human cancers via mutation of upstream negative regulators such as PTEN or TSC2. Following mTOR activation, DEPTOR is phosphorylated by mTOR, which in turn leads to phosphorylation of DEPTOR by CK1 (refs 26, 27). These phosphorylation events are recognized by SCFβ-TrCP ubiquitin ligase, resulting in DEPTOR polyubiquitination and proteolytic degradation26,27,28. Following its activation by mTORC1, p70S6K can also phosphorylate DEPTOR, thus facilitating β-TrCP binding28.
Herein we provide intriguing evidence for a novel oncogenic mechanism regulated by mutations of IDH1/2, whereby 2HG-induced KDM4A inhibition promotes mTOR activation. We show that KDM4A specifically interacts with DEPTOR, a negative regulator of both mTORC1 and mTORC2. KDM4A depletion/inactivation results in drastic reduction of DEPTOR levels, while KDM4A overexpression reduces DEPTOR ubiquitination by β-TrCP1. We also show that 2HG-mediated inhibition of KDM4A directly affects the stability of DEPTOR, leading to mTOR activation independently of the PI3K/AKT/TSC1-2 pathway. This newly revealed PTEN-independent mode of mTORC1/2 activation via the mutated IDH/2HG/KDM4A/DEPTOR pathway may provide an additional molecular mechanism to explain the oncogenic activity of IDH1/2 mutations in glioma and other cancers.
Results
IDH1/2 and PTEN mutations are mutually exclusive
Towards elucidating the oncogenic properties of mutated IDH1/2, we analysed The Cancer Genome Atlas (TCGA) and deduced that mutations in IDH1/2 and PTEN (Phosphatase and TENsin homologue), an important tumour suppressor frequently disrupted in many cancers, display mutual exclusivity in low-grade glioma (P<0.00001; Fisher’s exact test) and glioblastoma multiforme (P=0.005; Fisher's exact test; Fig. 1a). On the other hand, as previously reported29,30, mutations in the tumour suppressor tp53 often co-occur with IDH1/2 mutations in low-grade glioma (P<0.0001; Fisher's exact test) and glioblastoma multiforme (P<0.00001; Fisher's exact test) (Fig. 1a). Thus it appears that major cancer networks can be strongly influenced by IDH1/2 gene status. The mutual exclusivity between IDH1/2 and PTEN mutations suggests that abrogation of PTEN function might not confer a growth advantage to tumours carrying IDH1/2 mutations, allowing speculation that IDH1/2 mutations mimic PTEN loss or inactivation. PTEN governs multiple cellular functions such as survival, growth and energy metabolism by suppressing the phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway. Oncogenic activation of mTOR stimulates critical processes required for cancer cell survival, proliferation, angiogenesis and metastasis25. Importantly, even though no PTEN mutations were observed in IDH1/2-mutated gliomas, the mTOR pathway is nonetheless frequently activated in these tumours31.

(a) IDH1/2 and PTEN mutations are mutually exclusive in low-grade gliomas and glioblastomas. Data were obtained from The Cancer Genome Atlas (TCGA). Each vertical lane represents one patient; green lines depict the presence of a mutation. Statistical analysis was performed using the Fisher's exact test. (b) mTOR pathway activation in MEFs p53−/− cells expressing wt versus mutant IDH. Flag-tagged IDH was retrovirally transduced into MEFs p53−/− cells, and whole-cell lysates were harvested at 9 days post selection. Asterisk denotes a nonspecific band recognized by the antibody. (c) Intracellular accumulation of 2HG on IDH1/2 mutant expression as in b. 2HG levels were normalized against glutamate levels. Asterisks denote a statistical difference between IDH1/2 mutated cells and empty vector cells, two-sided t-test P<0.0001 (graph represents three independent experiments). Error bars represent standard deviation. (d) Increased cell size associated with mutated IDH1/2 expression. FSC measurement in FACS analysis demonstrates the shift in cell size in MEFs p53−/− cells expressing IDH1R123H (green line) or IDH2R172K (blue line) compared with empty vector-infected cells (black line). A total 250,000 cells were counted for each condition and graph is representative of four independent experiments. (e) Inhibition of mTORC1 rescues the increased cell size in IDH2R172K mutant cells. MEFs p53−/− cells expressing IDH2R172K or empty vector were treated with 25 nM rapamycin for 16 h before FACS analysis. A total 250,000 cells were counted for each condition. (f) Rapamycin or Torin1 treatment prevents mTORC1 activation in cells expressing IDH1/2 mutants. MEFs p53−/− cells expressing IDH1/2 mutants were treated with 100 nM rapamycin or 250 nM Torin1 for 24 h and whole-cell lysates blotted for markers of mTORC1 activation. (g) 2HG stimulates the mTOR pathway. MEFs p53−/− were treated with 2 mM of octyl-2HG for 4 h, and whole-cell extracts analysed by western blot. (h) Octyl-2HG treatment increases cell size. MEFs p53−/− cells were treated with 1 mM octyl-2HG for 8 h before FCS analysis by FACS. A total 25,000 cells were counted for each condition and graph is representative of two independent experiments.
IDH1/2 mutations and 2HG activate mTOR signalling
The above considerations prompted us to postulate the existence of a PTEN-independent mode of mTOR pathway activation by oncogenic IDH1/2, which might constitute an important determinant in glioma development. We first evaluated the possibility that the mTOR pathway is actually regulated by oncogenic IDH1/2. Murine embryonic fibroblasts (MEFs) retrovirally transduced with cancer-associated IDH1R132H or IDH2R172K, but not with wild-type counterparts, exhibited increased mTORC1 and mTORC2 signalling as highlighted by enhanced phosphorylation of p70S6K, S6 and Rictor, and AKT, respectively (Fig. 1b and Supplementary Fig. 1a). The extent of mTORC1 stimulation correlated with intracellular levels of the oncometabolite R-2-hydroxyglutarate (2HG) produced by mutant IDH1 and IDH2 (Fig. 1c). Intracellular accumulation of 2HG in mutated IDH1/2-expressing cells is not associated with decreased levels of αKG levels (Supplementary Fig. 1b), which has been shown, in different studies, to either inhibit32 or stimulate33,34,35 mTOR activity.
It has been shown that mTOR activity is a major determinant of cell size36. Therefore, to probe the physiological response to mutant IDH1/2-mediated activation of mTOR signalling, we compared the effects of IDH1WT, IDH1R132H, IDH2WT and IDH2R172K expression in MEFs on cell size using flow cytometry. Cells expressing mutated IDH1/2 were larger than control counterparts (Fig. 1d and Supplementary Fig. 1c,d), and this increase correlated with intracellular levels of 2HG (Fig. 1c). Moreover, treatment of IDH2R172K-expressing cells with the mTORC1 inhibitor rapamycin mitigated the effect on cell size (Fig. 1e). In addition, treatment with rapamycin or the mTORC1/2 inhibitor Torin1 abrogated S6 phosphorylation on serines 240 and 244 in IDH1/2 mutant-expressing cells, confirming the activation of mTORC1-dependent p70S6K by 2HG-producing mutants (Fig. 1f). Expression of IDH1R132H in cells treated with AGI-5198, an inhibitor of mutated IDH1 (ref. 37), failed to activate mTORC1 (Supplementary Fig. 1e), suggesting that oncogenic IDH1-mediated production of 2HG is responsible for mTOR stimulation.
To directly evaluate whether 2HG is responsible for activation of mTORC1/2, we synthesized cell permeable 1-octyl-D-2-hydroxyglutarate (octyl-2HG) (Supplementary Fig. 1f)38. Treatment of cells with octyl-2HG led to activation of mTORC1/2, as illustrated by increased phosphorylation of p70S6K, S6, Rictor and AKT in both MEFs and HeLa cells (Fig. 1g and Supplementary Fig. 1g) in a time- and dose-dependent fashion (Supplementary Fig. 1h,i) compared with the control. Furthermore, treatment with octyl-2HG caused a relative increase in cell size (Fig. 1h), consistent with activation of mTOR by 2HG. 2HG-mediated effects on phosphorylation of mTORC1 substrates and cell size were attenuated by rapamycin (Supplementary Fig. 1j,k), and effect on mTORC2 activity inhibited by Torin1 (Supplementary Fig. 1l). Taken together, our results demonstrate that IDH1/2 mutations engender an increase in 2HG levels that stimulate mTOR signalling via both mTORC1 and mTORC2.
KDM4A modulates mTOR activity
Previous studies showed that mutated IDH1/2-stimulated 2HG production inhibits the activity of αKG-dependent enzymes including (i) the Jumonji family of Fe(II)-dependent oxygenases comprising lysine demethylases and (ii) the TET family of 5-methylcytosine hydroxylases6,7. To evaluate the possibility that one or more enzymes among these families is/are responsible for regulation of mTORC1 activity, we performed a targeted short interfering RNA (siRNA) screen in HeLa cells using S6 phosphorylation on serines 240 and 244 as a readout of mTOR activity. Among the αKG-dependent enzymes tested, depletion of KDM4A showed the most potent increase in S6 phosphorylation (Fig. 2a). KDM4A is an Fe(II)- and αKG-dependent enzyme possessing a JmjC catalytic domain8,9, as well as tandem PHD- and tandem Tudor- domains which are thought to associate with methylated chromatin residues. KDM4A catalyses demethylation of di- and tri-methylated H3K9 and H3K36 (refs 9, 39, 40, 41), and modulates multiple cellular processes including the DNA damage response13, cellular senescence14,42,43, transcription40,44, DNA replication12,45 and innate immunity46. Knockdown of KDM4A using three different short hairpin RNAs (shRNAs) led to increased phosphorylation of the mTORC1 substrates p70S6KT389 and 4E-BP1S65, and of the mTORC2 substrate AKTS473 (Fig. 2b), thus validating our siRNA screening approach. As AKT modulates cell survival and depletion of KDM4A leads to AKT activation (Fig. 2b), we determined the function of KDM4A in cell survival on metabolic stress. KDM4A knockdown prevented cleavage of caspase-3 and PARP, two indicators of apoptosis, following serum withdrawal (Fig. 2c). Moreover KDM4A-depleted cells were larger than cells treated with siGFP, and treatment with the mTORC1 inhibitor rapamycin abrogated this phenotype (Fig. 2d). We emphasize that the entire KDM4 family shares common substrates; nonetheless KDM4A was the sole family member to influence mTORC1 activity, as depletion of KDM4B-D had no impact on phosphorylation of 4E-BP1, S6 and Rictor (Supplementary Fig. 2a,b). KDM4A is profoundly inhibited by 2HG, exhibiting an IC50 of 2.1 μM (Fig. 2e) as determined by RapidFire mass spectrometry. 2HG failed to further increase mTORC1 activity in KDM4A-depleted cells (Fig. 2f), suggesting that inhibition of KDM4A by 2HG-producing mutated IDH1/2 is responsible for mTORC1/2 activation.

(a) siRNA screen of αKG-dependent enzymes for the involvement in mTOR signalling in HeLa cells. The experiments were performed in triplicate and the results of all experiments were plotted. The levels of phosphorylated S6 on serines 240/244 were normalized to total S6 protein. (b) KDM4A knockdown increases mTORC1/2 signalling. Depletion of KDM4A by lentiviral infection in HeLa cells using three different shRNAs. Whole-cell lysates were blotted for mTORC1/2 activation. (c) Depletion of KDM4A protects against apoptosis induced by serum deprivation. HeLa cells expressing a control shRNA or shRNAs targeting KDM4A were grown in media containing the indicated concentrations of serum for 24 h. Cell lysates were then analysed by western blotting. (d) KDM4A depletion increases cell size in an mTORC1-dependent manner. MEFs p53−/− were treated with siKDM4A and, where indicated, with DMSO or 25 nM rapamycin, for 16 h before FSC measurements by FACS. A total 200,000 cells were counted for each condition. (e) Dose-dependent inhibition of KDM4A by R-2HG in in vitro demethylation assay (graph represents two independent experiments). (f) 2HG does not further stimulate mTORC1 in KDM4A-depleted HeLa cells. HeLa cells transfected with siGFP or siKDM4A were serum starved for 16 h before stimulation with 2 mM octyl-2HG for 4 h.
KDM4A interacts with mTORC1/2
To elucidate the molecular mechanism involved in the regulation of mTOR kinase activity by KDM4A, we evaluated whether the latter transcriptionally modulates either PTEN/AKT/mTOR pathway regulators or mTORC1/2 complex components. Depletion of KDM4A in HeLa cells revealed minor or no significant changes in mRNA levels for either PTEN or tuberous sclerosis 1/2 (TSC1/2) (Fig. 3a). TSC1/2 is a GTPase-activating protein complex that maintains Rheb (Ras homologue enriched in brain) in an inactive form, thereby causing mTOR inhibition47,48,49. In addition, mRNA levels of RHEB, mTOR, DEPTOR or the upstream activating kinase PDK1 were at most very modestly changed following KDM4A depletion (Fig. 3a). Similar observations were made at the protein level for KDM4A-depleted cells, where expression of PTEN, TSC2, mTOR, Rictor and components of the amino acid signalling arm (that is, Rag and LAMTOR) remained essentially unaltered (Supplementary Fig. 2c,d). Interestingly, KDM4A knockdown had no impact on phosphorylation of AKT at threonine 308, a site phosphorylated by PDK1 after PI3K activation, suggesting that activation of mTORC1 occurs downstream PDK1/AKT (Supplementary Fig. 2c). This was further confirmed by our result showing that pharmacological PI3K inhibition using CAL-101 did not block activation of mTORC2 upon depletion of KDM4A, as revealed by increased phosphorylation of AKTS473 (Supplementary Fig. 2e). Furthermore, knockdown of KDM4A in rapamycin-treated cells still caused activation of mTORC2, implying mTORC1-independent modulation of mTORC2 by KDM4A (Supplementary Fig. 2e).

(a) Relative mRNA levels of negative and positive regulators of the PTEN/AKT/mTOR pathway following KDM4A depletion. Quantifications of mRNAs by RT–qPCR were normalized against β-actin (ActB) mRNA. Asterisks denote a statistical difference between siKDM4A-treated cells and siGFP control cells, two-sided t-test P<0.05 (graph represents two independent experiences). Error bars represent standard deviation. (b) Co-immunoprecipitation of endogenous mTORC1/2 complex members with Flag-KDM4A in 293T transfected cells. (c) Comparison of mTORC1/2-associated proteins with Flag-tagged mTOR or KDM4A. The 293T cells were transfected with either Flag-eYFP, Flag-KDM4A or Flag-mTOR, and protein lysates were subjected to anti-Flag immunoprecipitation. (d) Co-immunoprecipitation of Flag-KDM4A and HA-DEPTOR in 293T cells. (e) Endogenous KDM4A co-immunoprecipitates with Flag-DEPTOR. (f) DEPTOR PDZ domain associates with endogenous KDM4A. Flag immunoprecipitation of flag-tagged full length or fragments of DEPTOR. The samples were not sonicated in this experiment to confirm that the interaction is independent of nucleus disruption. (g) Endogenous KDM4A and DEPTOR associate in 293E cells. (h) Endogenous KDM4A and DEPTOR co-immunoprecipitate in NHA-hTERT cells.
Since the control exerted by KDM4A on mTOR activity seems to occur downstream of PTEN–PDK1–AKT–TSC1/2, we investigated whether KDM4A might physically interact with the mTORC1/2 complexes. Consistent with this possibility, KDM4A co-immunoprecipitated numerous members of mTORC1 and mTORC2 including mTOR, Raptor, Rictor, mSin1 and DEPTOR (Fig. 3b and Supplementary Fig. 3a–c). We then qualitatively compared the amount of mTORC1/2 components interacting with mTOR or KDM4A. Flag-tagged KDM4A modestly interacted with mSin1 and Raptor when compared with the amount co-immunoprecipitated with mTOR. On the other hand, both KDM4A and mTOR immunoprecipitated similar amounts of DEPTOR (Fig. 3c and Supplementary Fig. 3d), suggesting that DEPTOR might represent the major mTORC1/2 component interacting with KDM4A. We confirmed the interaction between DEPTOR and KDM4A in cells co-expressing the exogenous tagged proteins (Fig. 3d). Moreover, ectopic Flag-tagged DEPTOR co-immunoprecipitated endogenous KDM4A (Fig. 3e) and this interaction appeared to be mediated by the DEPTOR PDZ domain (Fig. 3f). Individual depletion of mTOR or mSin1 did not alter the binding of KDM4A and DEPTOR (Supplementary Fig. 3e,f), supporting our hypothesis that DEPTOR is the major mTORC1/2 component interacting with KDM4A. Endogenous KDM4A and DEPTOR are also associated in 293E (Fig. 3g), HeLa (Supplementary Fig. 3g), as well as in glioma-related central nervous system-derived cells, that is, immortalized normal human astrocytes (NHAs; Fig. 3h). Surprisingly, endogenous KDM4B also interacted with DEPTOR (Supplementary Fig. 3h), but was unable to modulate mTOR activity (Supplementary Fig. 2a). Recombinant KDM4A and DEPTOR proteins purified from E.coli failed to interact (Supplementary Fig. 3i), suggesting that KDM4A–DEPTOR interaction might be indirect, or requires either post-translational modifications absent in bacteria or conformational changes in DEPTOR occuring during its association with mTORC1 or mTORC2. We performed a fractionation assay to confirm that KDM4A and DEPTOR localize to the same sub-cellular compartments. While DEPTOR is mostly cytoplasmic, KDM4A was present in both the nucleus and cytoplasm (Supplementary Fig. 4a,b).
KDM4A stabilizes DEPTOR
The stability of DEPTOR, an endogenous negative regulator of mTOR activity24, is tightly regulated in response to growth signals via mTOR in conjunction with the SCF(βTrCP) E3 ubiquitin ligase complex26,27,28. We sought to determine the impact of KDM4A on DEPTOR protein levels. KDM4A knockdown led to a significant decrease in both DEPTOR protein levels (Fig. 4a) and half-life after serum stimulation (Supplementary Fig. 5a-b). Transient expression of wild-type KDM4A or tandem Tudor-disrupted KDM4AD939R, but not of catalytically inactive KDM4AH188A, in KDM4A-depleted cells partially restored DEPTOR levels (Supplementary Fig. 5c,d). This partial rescue might reflect reduced transfection efficiency (40–50%) of lentiviral-transduced cells under our experimental conditions. It is also possible that the transient nature of KDM4A re-introduction may not allow enough time for complete restoration of DEPTOR levels. We cannot exclude, however, that a portion of the effects we observed on DEPTOR levels are independent of KDM4A and maybe due to shRNA off-targeting effects. In addition, overexpression of wild-type KDM4A significantly abrogated in vivo ubiquitination of DEPTOR by β−TrCP1 (Fig. 4b,c); moreover, this reduction was dependent on the catalytic activity of the demethylase, but not on its chromatin-binding ability, since overexpression of the KDM4AD939R Tudor domain mutant, but not of the KDM4AH188A catalytic dead mutant, decreased DEPTOR poly-ubiquitination (Fig. 4b,c). In addition, KDM4A interacted with β-TrCP ubiquitin ligases (Supplementary Fig. 5e). Consistent with our finding that inhibition of KDM4A decreased DEPTOR protein levels, expression of the 2HG-producing IDH1R132H mutant also caused such a reduction (Fig. 4d). Treatment of human fibrosarcoma HT1080 cells bearing a heterozygous IDH1R132C mutation with the mutated-IDH1-specific inhibitor AGI-5198 (ref. 37) caused a marked decrease in mTORC1/2 activity. Moreover this was rescued upon KDM4A depletion (Fig. 4e and Supplementary Fig. 5f,g), showing that 2HG-mediated inhibition of KDM4A is indeed responsible for activation of mTOR. Our data raise the intriguing possibility that cancer therapeutic regimens designed to inhibit mutated IDH1/2 would be expected to reduce oncogenic mTOR activity.

(a) KDM4A depletion reduces DEPTOR levels. HeLa cells were transduced with lentivirus expressing sh_KDM4A.3, sh_KDM4A.5, sh_KDM4A.7 or sh_Control and whole-cell lysates were blotted to determine DEPTOR levels. (b) The demethylase activity of KDM4A is required to block β-TrCP1-dependent ubiquitination of DEPTOR in vivo. HA IP was performed on 293E cells transfected with the indicated plasmids. (c) Quantification of DEPTOR ubiquitination by β-TrCP1 in the presence or absence of wild-type KDM4A or mutants. DEPTOR ubiquitination was quantified in three independent experiments using ImageJ. Asterisks denote a statistical difference, two-sided t-test, *P<0.05; **P<0.01. Error bars represent standard deviation. NS, not significant, P>0.05. (d) IDH1R132H expression diminishes DEPTOR levels. Flag-IDH1R132H was introduced in p53−/− MEFs by retroviral transduction and whole-cell lysates immunoblotted. (e) Inhibition of endogenous mutated IDH1R132C decreases mTOR activity in a KDM4A-dependent manner. HT1080 cells were transduced with lentivirus expressing sh_KDM4A.3 or sh_Control. The cells were treated with 3 μM AGI-5198 twice every day for 5 days and whole-cell lysates blotted for markers of mTOR activation. (f) Expression of mutant IDH1 in NHA-hTERT increases mTOR signalling. IDH1WT-Flag or IDH1R132H genes were introduced in astrocytes through retroviral infection and cell lysates were blotted for mTORC2 activation. (g) IDH1R132H overexpression increases 2HG production in astrocytes. Intracellular 2HG levels were normalized to glutamate. Asterisk denotes a statistical difference between IDH1R132H and empty vector cells, two-sided t-test P<0.0001 (graph represents three independent experiments). Error bars represent standard deviation. (h) Treatment of NHA-hTERT with octyl-2HG activates mTOR signalling. The cells were treated for 8 h with 1 mM octyl-2HG. The experiment shown is a representative of triplicates. (i) Non-canonical mTOR activation in IDH1/2WT and IDH1R132H human astrocytomas. Scale bar, 50 μm. Representative samples from IDH1/2WT (n=3) and IDH1R132H (n=3) human astrocytomas are shown.
IDH1 mutation and 2HG regulate mTOR in human astrocytes
To evaluate the impact of IDH mutations on mTOR activation in a more physiological setting, we first retrovirally transduced immortalized NHAs with empty vector, wild-type IDH1 or IDH1R132H. NHAs expressing mutant IDH1 displayed higher levels of p-AKTS473 (Fig. 4f) associated with increased accumulation of intracellular 2HG (Fig. 4g), but did not manifest decreased αKG (Supplementary Fig. 5h). In addition, treatment of NHAs with octyl-2HG led to activation of mTORC1 and mTORC2, as indicated by increased p-S6S240/244 and p-AKTS473, respectively (Fig. 4h). Finally, octyl-2HG decreased DEPTOR levels in NHA, as compared with control (Supplementary Fig. 5i).
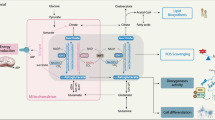
We then asked whether oncogenic IDH1 might be associated with markers of mTOR activation in human brain tumours. Immunohistochemistry analysis of IDH1R132H-mutated human-grade 2/3 astrocytoma samples revealed an increase in S6 phosphorylation on serines 240/244 (Fig. 4i). However, while 20 to 40% of the tumour cells from wild-type IDH1/2 astrocytomas showed strong staining for p-AKTT308, the upstream activator of mTORC1, less than 2% of IDH1R132H-mutated astrocytomas stained positively for p-AKTT308 (Fig. 4i). This suggests that activation of mTORC1 occurs downstream of the PI3K–PDK1–AKT cascade in tumours harbouring IDH1 mutations, supporting the notion that 2HG-mediated inhibition of KDM4A affects the stability of DEPTOR leading to mTOR activation independently of the PI3K/AKT/TSC1-2 pathway (Fig. 5).

Simplified schematic model for mTOR activation following KDM4A inhibition by IDH1/2 gain-of-function mutations.
Discussion
IDH mutations, frequently observed in brain tumours and leukaemia, lead to production of the oncometabolite 2HG, which in turn causes cell differentiation defects by impairing histone demethylation38. In addition, reversible inhibition of the 5′-methylcytosine hydroxylase TET2 by 2HG causes profound epigenetic changes contributing to transformation50,51. Herein we describe for the first time that oncogenic IDH and subsequent production of 2HG activate mTOR signalling. Either expression of mutated IDH1/2 or treatment with 2HG engendered activation of both mTORC1 and mTORC2 and, as a consequence, cell size increase. We note that the mTORC1 inhibitor rapamycin significantly, but only partially, decreased cell size of IDH2R172K-expressing cells. In mammalian systems, control of cell size by mTOR is mediated by both p70S6K and 4E-BP/eIF4E36. As these latter factors display differential sensitivity to rapamycin52, recovery of 4E-BP1 phosphorylation in rapamycin-treated IDH2R172K cells might explain the observed partial rescue of cell size (Fig. 1e).
Since 2HG constitutes an inhibitor of α-ketoglutarate-dependent enzymes, we used a targeted siRNA screen against these enzymes to identify KDM4A as a novel regulator of mTOR. Furthermore, our data indicate that KDM4A controls mTOR through modulation of DEPTOR stability. Indeed, KDM4A catalytic activity is essential for decreasing β-TrCP1-dependent poly-ubiquitination of DEPTOR (Fig. 4b,c) thereby promoting DEPTOR protein stability (Supplementary Fig. 5c). Further investigation will be required to identify the substrate(s) of KDM4A-mediated demethylation involved in controlling DEPTOR stability.
Amplification of KDM4A gene copy number, or of its mRNA expression, has been observed in various cancers. Such upregulation of KDM4A has been reported to promote site-specific genomic amplification45 or hinder p53 activation and senescence14. Conversely, genomic loss of KMD4A and concomitant decrease in mRNA levels has also been reported in multiple tumour types (Supplementary Fig. 6a–j)45, particularly bladder cancer where KDM4A downregulation correlates with tumour progression53. Our own results suggest that decreased KDM4A expression could contribute to mTOR activation during cancer development, suggesting that this demethylase could act as a context-dependent oncogene.
Recent evidence demonstrates that mTORC1 controls mitochondrial activity54 and stimulates metabolism of glutamine55, an amino acid catabolized into αKG, suggesting that mTOR modulates the TCA cycle. Here we provide evidence that TCA cycle defects can lead to aberrant activation of mTOR kinase. Indeed our data show that KDM4A associates with DEPTOR, and that 2HG-mediated inhibition of KDM4A in IDH1/2-mutated cells impacts DEPTOR protein stability leading to mTOR activation (Fig. 5). On the other hand, it has been reported that 2HG inhibits ATP synthase and mTOR activity in cell lines wherein mTOR is chronically hyperactivated due to the loss of PTEN activity or other causes56. Under such conditions, 2HG-mediated inhibition of ATP synthase might alter the ATP/ADP ratio leading to a decrease in mTOR activity. As a possible explanation for this controversy, we emphasize that our genomic analysis of human gliomas and glioblastomas revealed that IDH and PTEN mutations are mutually exclusive. In addition, our data show that 2HG stimulates mTORC1/2 activity in numerous cell lines where mTOR is not hyperactivated by abrogation of PTEN. Furthermore, mTOR signalling is readily observed in clinical samples of IDH1-mutated gliomas. This clearly suggests that the effects of 2HG on mTOR signalling rely on the integrity of upstream signalling pathways.
The novel PTEN-independent mode of mTORC1/2 activation via mutated IDH/2HG/KDM4A/DEPTOR revealed here may provide an additional molecular explanation for the oncogenic activity of IDH1/2 mutation in brain cancer. This would indicate a potential therapeutic strategy to target oncogenic mTOR signalling in cancers harbouring IDH1/2 mutations.
Methods
Cell culture
HEK293T, HeLa, HT1080 and 293E were obtained from the American Type Culture Collection (ATCC), p53−/− MEFs were obtained from Dr Gerardo Ferbeyre and previously described57, and normal human astrocytes immortalized by hTERT (NHA-hTERT) were kindly provided by Dr Nada Jabado. All the cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Hyclone), 1% sodium pyruvate (Gibco) and 1% penicillin/streptomycin (Gibco). Mycoplasma contamination check has been carried out for all cancer cell lines.
Plasmids and transfections
Cells were transfected with 10 μg of DNA using 30 μg of linear polyethylenimine. The following vectors were used: pcDNA3.1 Flag-eYFP, pLPC Flag-KDM4A, pCMV-Sport6-Flag-mTOR, pMSCV HA-DEPTOR, pDEST myc-KDM4A wild type or mutants (H188A and D939R), pcDNA3 Myc-β-TrCP1 (Addgene plasmid #20718; gift from Dr Yue Xiong) and pcDNA3 HA-Ub. The following plasmids, pRK5 Myc-mTOR (Addgene plasmid #1861), pRK5 Myc-Raptor (Addgene plasmid #1859), pRK5 Myc-Rictor (Addgene plasmid #11367), pRK5 Flag-DEPTOR (Addgene plasmid #21334), pRK5 Flag-DEPTOR DEP domains (Addgene plasmid #21700) and pRK5 Flag-DEPTOR PDZ domain (Addgene plasmid #21701) were gifts from Dr David Sabatini.
Lentiviral and retroviral infections
Lentiviral particles were generated by transfecting 293T cells using 5 μg of shRNA plasmid, 3.75 μg of psPAX2 packaging plasmid and 1.25 μg pMD2.G envelope plasmid. Media was changed 24 h after transfection and lentiviral particles were harvested 24 h later. Viral supernatant was filtered through 0.45 μm filters and supplemented with 8 μg ml−1 polybrene and 10% FBS. Viruses were titrated using limiting dilution in HeLa cells and the same MOI was used for each condition. The following plasmids were used: pLKO.1-sh_Control, pLKO.1-sh_KDM4A#3 (RHS3979-201745071, Dharmacon), pLKO.1-sh_KDM4A#5 (RHS3979-201745073, Dharmacon), pLKO.1-sh_KDM4A#7 (RHS3979-201745075, Dharmacon), pLKO.1-sh_mTOR#53 (TRCN0000195453, Sigma), pLKO.1-sh_mTOR#84 (TRCN0000039784, Sigma), pLKO.1-sh_mSin1#1 (13483, Addgene) and pLKO.1-sh_mSin1#2 (13484, Addgene).
Retroviruses were produced by transfecting packaging cells (Phoenix) with 10 μg of DNA using 30 μg of polyethylenimine. Media was changed 24 h later and viral supernatant harvested at 48 and 56 h post-transfection. Viral supernatants were filtered through 0.45 μm filters and supplemented with 8 μg ml−1 polybrene and 10% FBS. Media were changed 16 h after infection. Retroviral plasmids pLPCX IDH1WT Flag, pLPCX IDH1R132H Flag, pLPCX IDH2WT Flag and pLPCX IDH2R172K Flag are described in ref. 58. The pLXSN IDH1WT Flag and pLXSN IDH1R132H Flag were generated by PCR of the IDH1 genes (forward primer with EcoR1 site : 5′-TAGAATTCATGTCCAAAAAAATCAGTGG-3′, reverse primer with Xho1 site and Flag tag : 5′-ACCTCGAGTTACTTGTCATCGTCATCCTTGTAATCCATAAGTTTGGCCTG-3′). PCR products were inserted into the EcoRI- and XhoI-linearized pLXSN vector.
FACS-cell size determination
The cells were trypsinized and resuspended in phosphate-buffered saline (PBS) containing 2% FBS. Forward-scatter measurement (FSC) intensity was measured using BD FACSCalibur (BD Biosciences) and analysed with BD CellQuest Pro software (BD Biosciences). For each experiment 1,00,000–2,00,000 cells were analysed, except for cells treated with 2HG where 10,000–25,000 cells were used.
Immunoblots and antibodies
The cells were lysed in 10 mM Tris (pH 7.4), 120 mM NaCl, 0.5% Triton with protease inhibitor mix (cOmplete, Roche) and phosphatase inhibitors (PhosSTOP, Roche) and sonicated for 3 × 10 s. After centrifugation at 15,000g for 10 min, proteins were separated on SDS–PAGE and transferred to nitrocellulose or PVDF membranes (Bio-Rad). Primary antibodies used were anti-Flag (M2, Sigma, 1:5,000), anti-HA.11 (16B12, Covance, 1:1,000), anti-Myc (9E10, Sigma, 1:1,000), anti-α-Tubulin (B-5-1-2, Sigma, 1:10,000), anti-KDM4A (NB110-40585, Novus Biologicals, 1:1,000) and (N154/32, NeuroMab, 1:1,000), anti-mTOR (2972, Cell Signaling, 1:1,000) (7C10, Cell Signaling, 1:1,000), anti-DEPTOR (ABS222, Millipore, 1:1,000), anti-DEPTOR (D9F5, Cell Signaling, 1:500), anti-DEPTOR (NBP1-49674, Novus Biologicals, 1:1,000), anti-GβL (PA5-17442, Thermo Scientific, 1:1,000), anti-Raptor (24C12, Cell Signaling, 1:1,000), anti-phospho-Raptor S792 (2083, Cell Signaling, 1:1,000), anti-PRAS40 (PA5-17184, Thermo Scientific, 1:1,000), anti-Rictor (53A2, Cell Signaling, 1:1,000), anti-phospho-RictorT1135 (D30A3, Cell Signaling, 1:1,000), anti-mSin1 (07-2276, Millipore, 1:1,000), anti-Protor2 (D19A5, Cell Signaling, 1:1,000), anti-PTEN (A2B1, Santa Cruz, 1:1,000), anti-TSC2 (T.309-0 Thermo Scientific, 1:1,000), anti-RagA (D8B5, Cell Signaling, 1:1,000), anti-RagB (D18F3, Cell Signaling, 1:1,000), anti-RagC (D31G9, Cell Signaling, 1:1,000), anti-RagD (4470, Cell Signaling, 1:1,000), anti-LAMTOR1 (D11H6, Cell Signaling, 1:1,000), anti-LAMTOR2 (D7C10, Cell Signaling, 1:1,000), anti-LAMTOR3 (D38G5, Cell Signaling, 1:1,000), anti-p70S6K (9202, Cell Signaling, 1:1,000), anti-phospho-p70S6KT389 (108D2, Cell Signaling, 1:1,000), anti-4E-BP1 (53H11, Cell Signaling, 1:1,000), anti-phospho-4E-BP1S65 (9451, Cell Signaling, 1:1,000), anti-S6 (54D2, Cell Signaling, 1:1,000), anti-phospho-S6S240/244 (5364 and 2215, Cell Signaling, 1:1,000), anti-AKT PH domain (05-591, Millipore, 1:1,000), anti-phospho-AKTS473 (9271, Cell Signaling, 1:1,000) (D9E, Cell Signaling, 1:1,000), anti-phospho-AKTT308 (C31E5E, Cell Signaling, 1:1,000), anti-PARP (9542, Cell Signaling, 1:1,000), anti-Caspase 3 (9662, Cell Signaling, 1:1,000), anti-actin (D6A8, Cell Signaling, 1:2,000) and anti-GAPDH (D16H11, Cell Signaling, 1:2,000). Scans of ECL and/or X-ray films showing whole membranes are presented in Supplementary Fig. 7.
Immunoprecipitations
The cells were lysed in 1 ml of lysis buffer (0.3% CHAPS, 50 mM Hepes, 120 mM NaCl, 2 mM EDTA with protease inhibitor mix and phosphatase inhibitors (Roche)) and sonicated three times for 10 s each (except for Fig. 3f, for which samples were not sonicated to confirm that the interaction between KDM4A and DEPTOR occurred prior sonication). After centrifugation at 15,000g for 10 min, the supernatant was incubated for 2 h at 4 °C with 20 μl of Flag M2 agarose beads (Sigma). Immunoprecipitates were washed three times with lysis buffer containing 150 mM NaCl and twice with TBS before elution in Laemmli buffer.
For qualitative comparison of IP performed in CHAPS and Triton (Supplementary Fig. 3d), the cells were lysed in lysis buffer (40 mM Hepes pH 7.5, 120 mM NaCl, 1 mM EDTA with protease inhibitor mix Roche) containing either 0.25% CHAPS or 1% triton. The cells were then sonicated three times for 10 s each before centrifugation at 15,000g for 10 min. The supernatants were incubated for 1 h at 4 °C with 20 μl of Flag M2 agarose beads (Sigma). The immunoprecipitates were washed five times with lysis buffer and eluted in Laemmli buffer.
For HA-Ub IP, the cells were serum starved for 24 h followed by 30 min serum stimulation (10%) before addition of lysis buffer A (20 mM Tris pH 7.5, 1% Triton, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 20 mM N-ethylmaleimide with protease inhibitor mix and phosphatase inhibitors (Roche)). Sample were sonicated three times for 10 s each and immunoprecipitated with 20 μl of HA agarose beads (Sigma) for 2 h at 4 °C. The immunoprecipitates were washed three times with lysis buffer A before elution in Laemmli buffer.
For endogenous KDM4A IP, ∼2 × 108 cells (NHAs, HeLa or 293E) were resuspended in 5 ml of lysis buffer A with 0,5% triton. Extracts were prepared as described above and were pre-cleared for 1 h with 20 μl protA/G beads (Cell Signaling). Immunoprecipitations were performed with 12 μg of NO66 antibody (negative control, SCG consortium, ABC058-F03), KDM4A antibody (p014, SCG consortium) or KDM4B antibody (SCG consortium) overnight at 4 °C. 60 μl of M-280 streptavidin-coated dynabeads (Life Technologies) were added to each sample and incubated for 1 h at 4 °C. The beads were washed three times with lysis buffer A before elution in Laemmli buffer.
Production of recombinant proteins
The GST-DEPTOR (pGEX 5X3), His-KDM4A(aa 1-562) and His-KDM4A(aa 563-1064) (pDest17) were expressed in BL21 bacteria grown in LB. After induction with IPTG (0.2 mM) for 16 h at 20 °C, bacteria were harvested by centrifugation and lysed by sonication. The GST-fusion protein was purified on glutathione agarose beads (Sigma) and the His-fusion protein were purified with Ni-NTA agarose (Qiagen). The His-fusion proteins were eluted off the beads using 200 mM imidazole buffer and dialysed overnight against PBS, followed by concentration on Amicon columns (Millipore).
GST pull-down assays
For GST pull-downs, 1 μg of purified GST-DEPTOR recombinant protein was incubated for 2 h at 4 °C with 500 ng of His-KDM4A (aa 1–562) or (aa 563–1064) recombinant protein in GST pull-down buffer (25 mM HEPES pH7.5, 250 mM KCl, 0,1% NP-40, 10% glycerol, 100 μg ml−1 BSA). The beads were then washed with GST pull-down buffer four times and eluted in Laemmli buffer 2 × .
Metabolite extraction and quantification of 2HG
Quantification of citric acid cycle (CAC) organic acid intermediates was carried out at the Goodman Cancer Research Center metabolomics core facility (McGill University, Montreal, QC, Canada). Following cellular extraction and either keto-acid reduction or methoximation, GC/MS data were collected and analysed as described in detail59,60 except that 4 μl of the deuterated CAC standard mix was added due to the high level of 2-hydroxyglutaric acid in mutant IDH1/2-expressing cells. Resulting levels of CAC intermediates were normalized against cell number.
Synthesis of 1-octyl-D-2-hydroxyglutarate
Synthesis of 1-octyl-D-2-hydroxyglutarate was performed as previously described38. Briefly, commercial R(-)-tetrahydro-5-oxofuran-2-carboxylic acid (910 mg, 7 mmol) was dissolved in water (1 ml), cooled to 0 °C and treated with 1 N KOH (14.7 ml, 14.7 mmol). After stirring, the solution was dessicated under reduced pressure, co-evaporated with EtOH and dried. The white salt obtained was partially dissolved in trifluoroactic anhydride (50 ml) and the solution evaporated under reduced pressure. Anhydrous tetrahydrofuran (45 ml) was then used to dry and dissolve the residue, and octanol (2.3 ml, 14.7 mmol) was added to the resulting solution. After stirring overnight, water was added to quench the reaction and the mixture extracted with EtOAc. The extracts were combined, dried over MgSO4, concentrated and purified by flash chromatography eluting with Hexanes-EtOAc 3:1 to 1:1 giving 1-octyl-D-2-hydroxyglutarate (70 mg, 7 %) as an oil. 1H NMR (CD3OD) δ: 4.19 (m, 1H), 4.15 (m, 2H), 2.51–2.36 (m, 2H), 2.12–2.04 (m, 1H), 1.94–1.85 (m, 1H), 1.70–1.63 (m, 2H), 1.40–1.31 (m, 10H), 0.90 (t, 3H, J=6.8 Hz); MS (APCI neg) 259.05 [M-H]-.
siRNA screen targeting αKG-dependent oxygenases
HeLa cells were transfected using Lipofectamine RNAiMAX (Invitrogen) using reverse transfection protocol supplied by the manufacturer. Briefly, 20 nM siRNA duplexes and 10 μl RNAiMAX lipofectamine (Invitrogen) were diluted in 1 ml OPTI-MEM (Invitrogen) in 6 mm plates and incubated for 15 min before adding 600,000 cells per plate. Three days following the transfection, the cells were lysed in 10 mM Tris-HCl, 100 mM NaCl, 0.5% Triton X-100, pH 7.6 with protease inhibitor mix and phosphatase inhibitors (Roche), sonicated three times for 10 s, then centrifuged for 10 min at 10,000g. The supernatants were then analysed by western blotting for total S6 or phospho-S6S240/244, and quantification of band intensity was performed using ImageJ. All siRNAs were purchased from ThermoScientific (Dharmacon). For a complete list of siRNAs, see Supplementary Table 1.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde in PBS for 5 min at room temperature and permeabilized with 1% Triton X-100 in PBS for 5 min at room temperature. Fluorescent visualization of KDM4A was performed using pAb anti-KDM4A antibody (1:100, NB110-40585; Novus Biologicals) coupled with an Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L) secondary antibody (catalogue # 4412, Cell Signaling, 1:1,000). Polymerized actin was visualized using CF™568 phalloidin (catalogue # 00044-300U; Biotium). DNA was stained with 4′,6′-diamidino-2-phenylindole (DAPI). Immunofluorescent images were captured using a × 60 lens (1.42 NA, splan APO), mounted on an FV1000 confocal laser scanning microscope driven by FluoView software (Olympus). Image acquisition was performed at room temperature, the brightness/contrast tool in FluoView software (Olympus) was used to process images and montages were made using Photoshop CS6 (Adobe).
Cell fractionation
The harvested cells were suspended in cold extraction buffer (10 mM Hepes (pH 7,4), 5 mM MgCl2, 320 mM sucrose and 1% Triton X-100), agitated for 10 s and incubated on ice for 10 min. The cells were centrifuged at 2,000g and the supernatant (cytoplasmic fraction) collected while the pellet (nuclear fraction) was washed two times with 10 mM Hepes (pH 7,4) containing 5 mM MgCl2 and 320 mM sucrose.
Quantitative real-time PCR
Total RNA was isolated using Trizol (Invitrogen) according to the manufacturer's protocol. The High Capacity cDNA Reverse Transcription Kit was used with random primers (Applied Biosystems) to reverse transcribe 2 μg of total RNA in a final volume of 20 μl. GAPDH (glyceraldehyde-3-phosphate dehydrogenase), HPRT1 (hypoxanthine guanine phosphoribosyl transferase) and ACTB (Beta-actin) were used as endogenous controls and their gene expression levels determined using pre-validated Taqman Gene Expression Assays (Applied Biosystems). Quantitative PCR (qPCR) reactions were performed in 384-well plates using 1.5 μl of cDNA samples (5–25 ng), 5 μl of the TaqMan Fast Universal PCR Master Mix (Applied Biosystems), 0.5 μl of the TaqMan Gene Expression Assay (20X) and 3 μl of water in a final volume of 10 μl. The assays used to determine target gene expression levels were designed using the Universal Probe Library from Roche (www.universalprobelibrary.com). qPCR reactions for target genes were performed in 384-well plates using 1.5 μl of cDNA samples (5–25 ng), 5 μl of the TaqMan Fast Universal PCR Master Mix (Applied Biosystems), 2 μM of each primer and 1 μM of UPL probe in a final volume of 10 μl. Amplification levels were detected using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) programmed to run at 95 °C for 3 min before starting 40 cycles of 5 s at 95 °C and 30 s at 60 °C. The reactions were run in triplicate and the quantification performed using average Cts values. The ΔΔCT method was used to determine relative quantification of target genes. The primers used for RT–qPCR (qPCR with reverse transcription) are listed in Supplementary Table 2.
2-Hydroxyglutarate IC50 determination
The potency of 2-hydroxyglutarate for KDM4A inhibition was determined by RapidFire Mass Spectrometry61 and as previously described62. Briefly, 25 μl of recombinant human KDM4A (400 nM) was pre-incubated with 2-hydroxyglutarate during 15 min before starting the reaction by adding 25 μl of substrate mix (200 μM L-ascorbic acid, 20 μM Fe2+, 20 μM H3K9(me3) peptide and 20 μM 2-oxoglutarate). The enzymatic reaction was stopped after 50 min with 5 μl of 10% formic acid, then the reaction mixture was transferred to a RapidFire RF360 sampling robot. The samples were applied onto a C4 Solid Phase Extraction (SPE) cartridge by aspiration under vacuum. Non-volatile buffer components were removed from the C4 SPE by an aqueous wash step (0.1% formic acid in water). Then, the peptide was eluted from the C4 SPE onto an Agilent 6530 Q-TOF by an organic wash step (85% acetonitrile in water, 0.1% formic acid). Peak area data for the substrate and the product methyl marks were extracted from ion chromatograms and integrated using RapidFire Integrator software. The percentage of demethylation was calculated using Microsoft Excel software, and IC50 curves were generated by GraphPad Prism.
Protein stability assay
For half-life measurements, HeLa cells were starved for 24 h and pre-treated with 100 μg ml−1 cycloheximide and 40 μg ml−1 chloramphenicol 1 h before serum addition (10%). The cells were harvested at indicated time periods and whole cell lysates blotted. The protein levels were normalized to tubulin and quantified using ImageJ.
Immunohistochemical stainings of astrocytomas
Diffuse astrocytomas with IDH1R132H mutation (n=3) or wild-type IDH1/2 (n=3) were processed for immunohistochemistry. IDH1/2 wild-type status was confirmed by Sanger sequencing. Four-micrometre thick sections were cut from the selected formalin paraffin embedded specimens and stained using a Benchmark XT immunostainer (Ventana). The Ventana staining protocol included a pretreatment with Cell Conditioner 1 for 40 min, followed by incubation with the primary antibodies at 37 °C for 60 min. All the antibodies were diluted in the Ventana dilution buffer as followed: p-S6S240/244 (D68F8, Cell Signaling Technology, 1:1,000); p-AKT1T308 (Novus Biologicals, 1:30). Incubation was followed by detection using Optiview DAB IHC detection kit (Ventana).
Reproducibility of experiments
Representative figures were repeated as follows. Figs 1d,g; 2b,f and 3g,h and Supplementary Fig. 3g were reproduced at least four times. Figs 1b,c; 3b,f and 4a,b,g,i and Supplementary Figs 1b–d,k; 2b,c,e; 3a,c; 4a,b; 5a,c,g,h,i were repeated three times. Figs 1h and 2c,e, Figs 3a,c and 4d–f,h and Supplementary Figs 1a,g–i,l; 2d; 3b,d,i; 5e,f were reproduced twice.
Data availability
The TCGA data referenced in this study are available in a public repository from the TCGA website (http://cancergenome.nih.gov). The authors declare that all the other data supporting the findings of this study are available within the article and its Supplementary Information files, and from the corresponding authors upon reasonable request.
Additional information
How to cite this article: Carbonneau, M. et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 7:12700 doi: 10.1038/ncomms12700 (2016).
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Mardis, E. R. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 (2009).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Ward, P. S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Chowdhury, R. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 (2011).
Tsukada, Y. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Whetstine, J. R. et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467–481 (2006).
Katoh, M. & Katoh, M. Identification and characterization of JMJD2 family genes in silico. Int. J. Oncol. 24, 1623–1628 (2004).
Kim, J. et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 7, 397–403 (2006).
Black, J. C. et al. Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol. Cell 40, 736–748 (2010).
Mallette, F. A. et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31, 1865–1878 (2012).
Mallette, F. A. & Richard, S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep. 2, 1233–1243 (2012).
Hara, K. et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 (2002).
Kim, D. H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002).
Jacinto, E. et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128 (2004).
Sarbassov, D. D. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 (2004).
Sancak, Y. et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 (2007).
Vander Haar, E., Lee, S. I., Bandhakavi, S., Griffin, T. J. & Kim, D. H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323 (2007).
Frias, M. A. et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 16, 1865–1870 (2006).
Pearce, L. R. et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 405, 513–522 (2007).
Kim, D. H. et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 11, 895–904 (2003).
Peterson, T. R. et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 (2009).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Gao, D. et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303 (2011).
Duan, S. et al. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol. Cell 44, 317–324 (2011).
Zhao, Y., Xiong, X. & Sun, Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316 (2011).
Ohgaki, H. & Kleihues, P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol. 28, 177–183 (2011).
Liu, X. Y. et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 124, 615–625 (2012).
Pollack, I. F. et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv. Syst. 27, 87–94 (2011).
Chin, R. M. et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510, 397–401 (2014).
Duran, R. V. et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 47, 349–358 (2012).
Lorin, S. et al. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 9, 850–860 (2013).
Marino, G. et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 53, 710–725 (2014).
Fingar, D. C., Salama, S., Tsou, C., Harlow, E. & Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16, 1472–1487 (2002).
Rohle, D. et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 (2013).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Cloos, P. A. et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442, 307–311 (2006).
Klose, R. J. et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442, 312–316 (2006).
Shin, S. & Janknecht, R. Diversity within the JMJD2 histone demethylase family. Biochem. Biophys. Res. Commun. 353, 973–977 (2007).
Johmura, Y. et al. SCF(Fbxo22)-KDM4A targets methylated p53 for degradation and regulates senescence. Nat. Commun. 7, 10574 (2016).
Neault, M., Mallette, F. A. & Richard, S. miR-137 modulates a tumor suppressor network-inducing senescence in pancreatic cancer cells. Cell Rep. 14, 1966–1978 (2016).
Zhang, D., Yoon, H. G. & Wong, J. JMJD2A is a novel N-CoR-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2). Mol. Cell. Biol. 25, 6404–6414 (2005).
Black, J. C. et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154, 541–555 (2013).
Jin, J. et al. Noncanonical NF-kappaB pathway controls the production of type I interferons in antiviral innate immunity. Immunity 40, 342–354 (2014).
Garami, A. et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 (2003).
Inoki, K., Li, Y., Xu, T. & Guan, K. L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 (2003).
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C. & Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268 (2003).
Figueroa, M. E. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010).
Losman, J. A. et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 (2013).
Choo, A. Y., Yoon, S. O., Kim, S. G., Roux, P. P. & Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl Acad. Sci. USA 105, 17414–17419 (2008).
Kauffman, E. C. et al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol. Carcinog. 50, 931–944 (2011).
Morita, M. et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711 (2013).
Csibi, A. et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854 (2013).
Fu, X. et al. 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 22, 508–515 (2015).
Mallette, F. A. et al. Transcriptome analysis and tumor suppressor requirements of STAT5-induced senescence. Ann. N. Y. Acad. Sci. 1197, 142–151 (2010).
Li, S. et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 15, 57–68 (2013).
Mamer, O. et al. The complete targeted profile of the organic acid intermediates of the citric acid cycle using a single stable isotope dilution analysis, sodium borodeuteride reduction and selected ion monitoring GC/MS. Metabolomics 9, 1019–1030 (2013).
McGuirk, S. et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 1, 22 (2013).
Hutchinson, S. E. et al. Enabling lead discovery for histone lysine demethylases by high-throughput RapidFire mass spectrometry. J. Biomol. Screen. 17, 39–48 (2012).
Ng, S. S. et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 448, 87–91 (2007).
Acknowledgements
We thank (i) Drs Albert Lai, Nada Jabado and Philippe P. Roux for sharing reagents; (ii) Drs Elliot Drobetsky, Florence Couteau, Gerardo Ferbeyre and El Bachir Affar for providing reagents, fruitful discussions and critical reading of the manuscript; (iii) Mathieu Neault for artwork; (iv) the organic synthesis service of the Centre de Recherche du Centre Hospitalier Universitaire de Québec (CRCHUQ, Québec, Canada) for synthesis of 1-octyl-D-2-hydroxyglutarate; (v) Raphaëlle Lambert and the Genomics core facility of the Institute for Research in Immunology and Cancer (IRIC, Montréal, Canada); and (vi) Daina Avizonis (Rosalind and Morris Goodman Cancer Research Centre Metabolomics Core Facility; supported by the Canada Foundation of Innovation, The Dr John R. and Clara M. Fraser Memorial Trust, Terry Fox Foundation Team Grant #116128, and McGill University) for metabolite measurements. This study was supported by grants from the Fondation de l’Hôpital Maisonneuve-Rosemont (to F.A.M.), the Cole Foundation (to F.A.M.), the Canadian Institutes of Health Research (CIHR, MOP-133442 to F.A.M.; MOP-286437 to M.-E.H.), Natural Sciences and Engineering Research Council of Canada (NSERC, #402880-2012 to M.-E.H.), the Arthritis Research UK (program grant number 20522), the NIHR Oxford Biomedical Research Unit and the Rosetrees Trust to U.O. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from Abbvie, Bayer Healthcare, Boehringer Ingelheim, CIHR, the Canadian Foundation for Innovation, Eli Lilly and Company, Genome Canada, GlaxoSmithKline, the Ontario Ministry of Economic Development and Innovation, Janssen, the Novartis Research Foundation, Pfizer, Takeda and the Wellcome Trust. A.M. is recipient of Ph.D. studentships from the Cole Foundation and the Fonds de Recherche du Québec - Santé (FRQS)/Fondation des Étoiles. M.-E.L. received a post-doctoral fellowship from the CIHR. M.-E.H. and F.A.M. are Junior 1 Research Scholars of the FRQS. F.A.M. holds a Canada Research Chair in Epigenetics of Aging and Cancer.
Author information
Authors and Affiliations
Contributions
M.C., M.-E.H. and F.A.M. designed the study. M.C. performed most of the experiments, M.-A.G. performed the siRNA screen and treatments with 2HG and AGI-5198. L.M.G. performed the experiments with the PI(3)K inhibitor and AGI-5198, as well as determined DEPTOR levels upon KDM4A depletion. M.-E.L. performed endogenous co-immunoprecipitations. B.S. and M.L. carried out cell survival experiments, and provided tools and reagents. A.M., L.H., I.T. performed and analysed cell size by FACS. A.T. and U.O. performed and analysed in vitro demethylation assays. E.E.V. and R.G.J. performed metabolomic analyses. M.-C.G. and K.P. carried out immunohistochemistry on human gliomas and performed the histopathological analysis. M.C., M.-A.G., M.-E.H. and F.A.M. wrote the paper. All the authors discussed the results and commented on the manuscript at all the stages.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-7, Supplementary Table 1-2 and Supplementary References (PDF 6413 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Carbonneau, M., M. Gagné, L., Lalonde, ME. et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun 7, 12700 (2016). https://doi.org/10.1038/ncomms12700
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms12700
This article is cited by
-
Characterization of purinergic signaling in tumor-infiltrating lymphocytes from lower- and high-grade gliomas
Purinergic Signalling (2024)
-
Both Enantiomers of 2-Hydroxyglutarate Modulate the Metabolism of Cultured Human Neuroblastoma Cells
Neurochemical Research (2024)
-
L-2hydroxyglutaric acid rewires amino acid metabolism in colorectal cancer via the mTOR-ATF4 axis
Oncogene (2023)
-
Signaling pathways in brain tumors and therapeutic interventions
Signal Transduction and Targeted Therapy (2023)
-
Iron drives anabolic metabolism through active histone demethylation and mTORC1
Nature Cell Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
