
Abstract
Single-atom catalysts maximize the utilization of supported precious metals by exposing every single metal atom to reactants. To avoid sintering and deactivation at realistic reaction conditions, single metal atoms are stabilized by specific adsorption sites on catalyst substrates. Here we show by combining photoelectron spectroscopy, scanning tunnelling microscopy and density functional theory calculations that Pt single atoms on ceria are stabilized by the most ubiquitous defects on solid surfaces—monoatomic step edges. Pt segregation at steps leads to stable dispersions of single Pt2+ ions in planar PtO4 moieties incorporating excess O atoms and contributing to oxygen storage capacity of ceria. We experimentally control the step density on our samples, to maximize the coverage of monodispersed Pt2+ and demonstrate that step engineering and step decoration represent effective strategies for understanding and design of new single-atom catalysts.
Similar content being viewed by others


Introduction
Single-atom catalysts represent the limiting realization of supported metal catalysts with metal load ultimately dispersed as single atoms1,2. This maximizes the utilization of supported metals and helps development of sustainable catalytic technologies for renewable energies and environmental applications with reduced precious metal contents3,4. A central prerequisite for understanding and knowledge-based design of single-atom catalysts is the identification of specific adsorption sites on catalyst supports that provide the stabilization of single metal atoms under reaction conditions at elevated temperatures and pressures. For oxide supports, understanding specific adsorption sites presently concentrates on low-index oxide facets5,6,7,8,9. Single-atom catalysts are, however, nanostructured large-area materials; thus, a question arises whether single supported atoms can be stabilized at defect sites of nanostructured oxide supports.
Highly dispersed platinum (Pt) ions on ceria qualify as single-atom catalysts2 and hold a promise of radical reduction of Pt load in critical large-scale catalytic applications—hydrogen production3, three-way catalytic converters10 and fuel cells11. Ceria surfaces provide a limited amount of low coordinated surface sites where Pt2+ ions can adsorb and remain stable in real applications2,3,10,11. Recent studies on large-area ceria samples identify the necessity of nanostructuring the ceria substrates for obtaining supported Pt2+ ions2,4 and propose a square-planar PtO4 unit as a Pt2+-containing surface moiety4. In the present model study on the single crystalline CeO2(111) surface, we demonstrate that single-ion dispersions of Pt2+ are stabilized at monolayer (ML)-high ceria step edges. Pt2+ ions at step edges are located in PtO4 units that can be considered the elementary building blocks of Pt2+/ceria single-atom catalysts. The PtO4 units incorporate excess O and can act as oxygen source for redox reactions. Besides clarifying the nature of Pt2+ stabilization on ceria, our study demonstrates the importance of step edges—the most common surface defects on oxide supports12—for single-atom catalyst stabilization. We experimentally adjust the step density on the ceria supports for maximizing the load of monodispersed Pt2+ ions. This identifies step engineering13 and step decoration14,15 as advanced techniques for designing new single-atom catalysts.
Results
Pt deposits on highly defined CeO2(111) surfaces
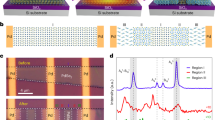
The experiments were performed on model CeO2(111) surfaces prepared as 20 to 40 Å thick ceria films on Cu(111) using procedures that allow adjusting the density of ML-high steps16 and the density of surface oxygen vacancies on ceria surface17. On these highly defined surfaces, we deposit 0.06 ML of Pt, anneal at 700 K in ultra-high vacuum (UHV) and observe stabilization of Pt2+ species and/or nucleation of Pt clusters with scanning tunnelling microscopy (STM) and with photoelectron spectroscopy (PES). Deposition and annealing of Pt on CeO2(111) surfaces containing low concentrations of defects—ML-high steps and surface oxygen vacancies (Fig. 1a)—yield metallic Pt0 clusters (Fig. 1b,c) coexisting with ionic Pt2+ species (Fig. 1c). To determine whether the charge of the supported Pt species is selectively induced by a specific defect type, we repeat the experiment varying independently the amount of surface O vacancies—up to 0.16 ML, creating CeO1.7 surface (Fig. 1d–f)—and the amount of ML-high steps on the CeO2(111) surface—up to 0.15 ML (Fig. 1g–i). We observe that surface oxygen vacancies do not promote the dispersion of Pt2+ species but lead to small metallic Pt0 clusters (Fig. 1e,f)18. On the other hand, the increased step density leads to almost complete oxidation of the Pt deposit to Pt2+ (Fig. 1h,i) proving that step edges selectively promote the stabilization of Pt2+ species. Detailed STM images allow to exclude formation of three-dimensional and two-dimensional PtOx clusters (Supplementary Fig. 1), and allow to conclude that Pt2+ species are incorporated in the ceria step edges. Nucleation of Pt0 clusters and stabilization of Pt2+ species represent concurrent processes. Differently to Pt0 clusters (Fig. 1b,e), Pt2+ species at the step edges are not discernible in empty states STM imaging (cf. Fig. 1g,h without and with Pt deposit) because of their electronic structure. STM imaging in occupied states on metal-supported ceria is unavailable19.

(a–c) CeO2(111) surface with low density of surface oxygen vacancies and ML-high steps. (d–f) CeO1.7 surface with increased density of surface oxygen vacancies. (g–i) CeO2(111) surface with increased density of ML-high steps. (a,d,g) STM images of clean surfaces before deposition of Pt. (b,e,h) STM images after deposition of 0.06 ML Pt and annealing at 700 K in UHV. All STM images 45 × 45 nm2, tunnelling current 25–75 pA, sample bias voltage 2.5–3.5 V. Scale bar, 20 nm (a). (c,f,i) PES spectra of the Pt deposit after annealing. All PES spectra were acquired with photon energy hν=180 eV (black points). Fits indicate metallic (Pt0, blue line) and ionic (Pt2+, red line) contributions to Pt 4f signal. EB is the photoelectron binding energy.
The possibility to adjust the density of ML-high steps on the model CeO2(111) surfaces16 allows us to obtain a quantitative correlation between the step density and the amount of Pt2+ species. We prepare CeO2(111) samples with step density between 0.06 and 0.20 ML16 and deposit 0.06 or 0.18 ML Pt at 300 K. Parameters of the prepared samples are summarized in Supplementary Table 1. After annealing at 700 K the amount of Pt stabilized in the form of Pt2+ is determined by PES. For quantification, all relevant parameters—the density of ceria steps, deposited amount of Pt and amount of stabilized Pt2+—are expressed in ML where 1 ML corresponds to the density of Ce atoms on the CeO2(111) surface, that is, 7.9 × 1014 cm−2. The density of steps is defined as the density of Ce atoms located at the ceria step edges20.
The amount of Pt2+ ions as a function of the step density is plotted in Fig. 2a. For the higher Pt coverage 0.18 ML, the analysis reveals a linear dependence between the amount of stabilized Pt2+ ions and the step density (Fig. 2a, blue symbols), confirming the activation of Pt oxidation to Pt2+ and the localization of Pt2+ at the surface steps. The highest step density 0.20 ML allows converting up to 80% of the Pt deposit to Pt2+. The degree of oxidation of the Pt deposit increases with decreasing the amount of deposited Pt to 0.06 ML (Fig. 2a, black symbols). In this case, up to 90% of Pt converts to Pt2+. The concentrations of ceria step edges and the amount of Pt2+ stabilized on the surface obey a classical supply-and-demand scenario characteristic for single-atom catalysts1,4,10: when sufficient step edges, the amount of oxidized Pt2+ is limited by the amount of deposited Pt. Otherwise, the amount of oxidized Pt2+ is limited by the step density regardless of the amount of deposited Pt. The Pt deposit exceeding the available step sites cannot be oxidized and nucleates as metallic Pt0 clusters on the surface. Besides the high oxidative power of the step edges towards Pt, our quantitative analysis reveals also the capacity of ceria step edges to accommodate a high density of Pt2+ ions. Up to 0.16 ML of Pt2+ ions can be stabilized by the sample containing 0.18 ML of Pt deposit and 0.20 ML of steps (Fig. 2a, blue symbols). This corresponds to 80% of the step-edge sites being occupied by Pt2+. In the whole range of the step densities between 0.06 and 0.20 ML, the occupation of the step-edge sites by Pt2+ varies between 50 and 80%.

(a) Amount of Pt2+ stabilized on CeO2(111) substrates with different density of steps for 0.18 ML (blue symbols) and 0.06 ML (black symbols) of deposited platinum. Pt not stabilized in the form of Pt2+ remains metallic. Lines represent guides to the eyes. Blue line is a linear fit of 0.18 ML Pt data. Black line represents the maximum achievable amount of Pt2+ in the case of 100% oxidation of Pt for 0.06 ML of deposited Pt. (b) Reduction of the ceria surface accompanying the stabilization of Pt2+ ions determined by resonant PES expressed as a coverage of the surface by Ce3+ ions. Lines represent guides to the eyes from a and indicate the Pt2+ concentration. The Ce3+ concentration is lower or equal to the Pt2+ concentration on all samples.
Stability and charge state of Pt2+/CeO2(111) samples
The necessity of annealing the Pt deposit on ceria in UHV at 700 K for obtaining Pt2+ stabilization in our experiment indicates the activated nature of Pt segregation at the ceria steps and oxidation, and implies considerable thermal stability of Pt2+ ions on ceria. High-temperature annealing represents a prerequisite for obtaining Pt2+ ions also in the experiments on large-area nanostructured ceria samples3,4. Once created, Pt2+ ions remain stable on repeated annealing at 700 K in UHV. The Pt2+ ions in our experiment also remain stable on adsorption and thermal desorption of CO in UHV (Supplementary Fig. 2), or on exposure to air at ambient conditions (Supplementary Fig. 3).
Parallel to the charge state of the Pt deposit we determine the charge state of the CeO2 support, in particular the concentration of surface Ce3+ ions that is indicative of reduction of the ceria surface. Contrary to the case of stabilizing Ni2+ ions on ceria21, we observe that Pt oxidation during annealing is not accompanied by a corresponding reduction of CeO2(111) surface (Fig. 2b). This rules out the direct participation of ceria into the observed Pt oxidation at steps and indicates the involvement of other oxidizing agents in the Pt2+ stabilization, such as excess oxygen atoms. In the UHV environment of our experiments, the eligible source of excess oxygen can be water adsorbing in sub-ML amounts from background atmosphere (Supplementary Fig. 4) and undergoing dissociation on reduced ceria and Pt/ceria substrates22,23. In the large-area Pt2+/CeO2 catalysts displaying high concentration of Pt2+ ions and exceptional redox reactivity, excess O atoms may be incorporated during the synthesis that proceeds in air3,10.
Segregation of Pt at CeO2(111) step edges
Ab initio density functional theory (DFT) calculations allow to interpret the above experimental results. We calculate the segregation thermodynamics and the atomic and electronic structures of Pt atoms in representative adsorption sites on CeO2(111) surfaces. The results for the lowest-energy configurations are summarized in Fig. 3 and Table 1. The model adsorption sites include oxygen vacancies (Fig. 3a), regular sites (Fig. 3b) and Pt clusters24 (Fig. 3c) on the CeO2(111) terrace, as well as two low-energy ML-high steps, which we label following ref. 25 as step I (Fig. 3d) and step II (Fig. 3e). A detailed list of the systems considered in the DFT analysis is reported in the Supplementary Note 1. The steps I and II represent the preferred types of steps at the CeO2(111) surfaces at temperatures <1,000 K (ref. 25). On our experimental samples, the steps I and II appear in equal proportion as evidenced from the absence of triangularly shaped islands in Fig. 1a,g25,26.

(a) Pt adatom in a surface O vacancy, (b) on the stoichiometric CeO2(111) terrace and (c) supported Pt6 cluster. (d) Pt adatom at the stoichiometric step I (step I-S) and (e) at the stoichiometric step II (step II-S). (f) Pt adatom at the step I with excess O (step I-O) and (g) at the step II with excess O (step II-O). Binding energies and Bader charges are summarized in Table 1. (d–g) The * symbol denotes the O atom removed to calculate the O vacancy formation energy reported in Table 2.
In agreement with the experiment, our calculations predict the preferential segregation of Pt adatoms at the steps I and II, independently on the local step geometry and stoichiometry. The binding energies of Pt at the steps are 1.6–3.4 eV higher than at stoichiometric or defective (111) terraces (Table 1). This driving force for Pt segregation at the steps is in qualitative agreement with recent calorimetric studies of other metal clusters on CeO2(111)27,28, see the Supplementary Discussion. The particular binding energy and the charge state of Pt atoms at the steps depend on both the local step geometry and stoichiometry (Table 1). In the following, we show that a good agreement with all the experimental observations can only be achieved when considering segregation of Pt at steps in the presence of excess O (calculations denoted as O), while Pt segregation at stoichiometric steps (denoted S) exhibits significant discrepancies.
Pt at stoichiometric CeO2(111) step edges
Pt segregation on step I-S yields Pt2+ species that are coordinated by four lattice O atoms in a characteristic PtO4 planar unit (Fig. 3d). The PtO4 unit is remarkably similar to that one proposed for Pt-doped ceria nanoparticles4 and for surface reconstructions of Pd–ceria systems29. Instead, the different atomic structure of the step II-S edge prevents the formation of PtO4 units, hinders the full Pt oxidation to Pt2+ and yields weakly oxidized PtΔ+ species (Fig. 3e). Calculation results presented in Fig. 3d–g correspond to the Pt coverage at the steps 1/3 (1 Pt atom per 3 Ce step-edge atoms). For interpreting the capacity of the ceria step edges to accommodate a high density of Pt2+ ions, we calculate the adsorption of Pt at the ceria steps with increasing Pt coverage at the steps (Fig. 4), ranging from 1/3 to 1 (1 Pt atom per 1 Ce step-edge atom). On the step I-S, the maximum coverage of Pt2+ species is 2/3 (Fig. 4a). Higher Pt2+ coverages are unattainable and lead to nucleation of metallic Pt clusters, due to the large strain buildup resulting from long sequences of interconnected PtO4 step units (Fig. 4b). On the step II-S, metallic Pt0 species appear already for a coverage higher than 1/3 (Fig. 4c). Thus, on samples with equal proportion of the stoichiometric steps I and II, ab initio calculations predict maximum Pt2+ coverage at the steps (≤33% of the step-edge sites) and maximum conversion of the Pt deposit to Pt2+ (≤33% of deposited Pt) that are well below the experimental values (50–80% of step-edge sites, up to 90% of deposited Pt, cf. Fig. 2a).

Calculated top views of the Pt binding to the steps I-S (a,b), step II-S (c), step I-O (d,e) and the step II-O (f,g) for Pt step coverage 2/3 (a,c,d,f) and 1 (b,e,g). At the step I-S, the limiting coverage of Pt2+ is 2/3 (a), additional Pt attaches to step edge as Pt0 (b). At the step II-S, the Pt2+ coverage is 0. Pt atoms attach as weakly ionized PtΔ+ and readily form metallic dimers (c) and clusters. On both steps I-O and II-O, excess oxygen can stabilize ionic Pt2+ at step edges as single ions appearing isolated or in groups up to 100% step coverage (d–g). The * symbol denotes the O atom removed to calculate the O vacancy formation energy reported in Table 2.
Most importantly, the calculations on the stoichiometric steps predict that Pt segregation, oxidation and the formation of the Pt2+ species are always accompanied by the reduction of surface Ce atoms from Ce4+ to Ce3+ (denoted in gray in Figs 3 and 4). The resulting concentration of the Ce3+ ions exceeds that of the Pt2+ ions by a factor of 2. This is in stark contrast with the resonant PES measurements on our samples showing that the concentration of Ce3+ is considerably lower than the concentration of Pt2+ after annealing the samples (Fig. 2b). This indicates that Pt is preferentially oxidized by other mechanisms than the Pt0/Ce4+ redox couple.
Pt2+ at CeO2(111) step edges with excess O
Agreement between the theory and the experiment can be achieved when taking into account the step edges in the presence of an excess of O atoms. Irrespective of the local step geometry and Pt coverage at the steps, we find that excess O atoms readily bind to Pt at the ceria steps and drive a rearrangement of the step morphology forming ionized Pt2+ species incorporated in the planar PtO4 moieties on both steps I and II (Fig. 3f,g). In the presence of excess of oxygen, Pt atoms bind stronger to the ceria step edges, with calculated binding energies up to 6.7 eV, which are higher than at the stoichiometric steps edges by ∼1.6 eV, and which are also higher than the cohesive energy of bulk metallic Pt (Table 1). This condition, which determines the stability of the Pt2+ species at steps with respect to metallic Pt clusters, is fulfilled only in the presence of excess oxygen at the steps. The computed electronic structure and density of states of the PtO4 moieties at the steps I-O and II-O (Supplementary Figs 5 and 6) confirm that the Pt0→Pt2+ oxidation results from the ionic Pt–O bond in the PtO4 planar units, and that Ce3+ ions do not form in agreement with the experimental evidence (Fig. 2b). The calculated maximum coverage of Pt2+ at the steps I-O and II-O is 100% (Fig. 4e,g and Supplementary Table 2), as interconnected assemblies of the PtO4 units can optimally fit the periodicities of both steps I and II at calculated Pt coverages at the step edges 1/3, 2/3 and 1 (Figs 3f,g and 4d–g). The presence of excess oxygen at steps therefore explains also the maximal Pt2+ ionization experimentally measured on the ceria-supported catalysts.
The stabilization of excess oxygen in the PtO4 moieties by the Pt2+ ions suggests an oxygen source for redox reactions and hence provides a link between the presence of highly dispersed ionic Pt species on ceria and the increased redox reactivity of Pt2+/CeO2 single-atom catalysts3. The oxygen buffering capacity of ceria-based catalysts is associated with easy oxygen vacancy formation. We calculate the vacancy formation energy on the clean CeO2(111) terrace, on the stoichiometric steps and on the Pt-decorated steps (Table 2). Compared with the energy of 2.5 eV calculated on the CeO2(111) terrace, the energies required to remove an oxygen atom bound to Pt at the stoichiometric steps are 3.3 eV (from the PtO4 unit at the step I-S) and 3.8 eV (from the PtO4 unit at the step II-S; see Table 2). Pt segregation at the stoichiometric step edges yields the formation of strong Pt–O bonds and therefore hinders ceria O-buffering. Much lower energies are instead needed to remove the excess O incorporated in the PtO4 units at steps I-O and II-O, where the O vacancy formation energy can be as low as 1.7 eV, lower or comparable to the values for the stoichiometric steps without Pt (2.0–1.8 eV; Table 2)30. This indicates that the dispersed Pt2+ ions can enhance the oxygen storage capacity of ceria-based catalysts by assisting the reversible storage of excess O atoms.
The single-ion nature of Pt2+ in the PtO4 units is preserved at all coverages, even when densely packed at the ceria step edges as interconnected PtO4 units. Indeed, the Pt charge state, its local electronic structure and the O vacancy formation energy of the densely packed PtO4 units are comparable to that of the isolated PtO4 units at the steps (Supplementary Fig. 7 and Table 2). The PtO4 units exhibit a large adaptability in stabilizing at different types of surface step edges, resulting in high effectiveness and capacity of the ceria surface to accommodate the Pt2+ ions. Regardless of the particular organization on the surface, the PtO4 units are also always accessible to the reactants. Thus, the square-planar PtO4 units carrying monodispersed Pt2+ ions can be considered elementary building blocks of single-atom Pt-ceria catalysts.
Discussion
On large-area samples, Pt2+ ions on ceria show exceptional reactivity with minimum Pt load in important applications: water–gas shift reaction3, hydrogen oxidation on the anode of proton-exchange membrane fuel cell11 and in the three-way catalyst converter10. In these applications, Pt2+ ions on ceria exhibit long-term stability under realistic reaction conditions of elevated temperatures and ambient pressure of reactant gases3,31,32. Pt2+ in large-area samples is routinely identified with PES. Complementary measurements with extended X-ray absorption fine structure (EXAFS)33 and high-resolution transmission electron microscopy3,4,31,32 confirm the absence of three-dimensional Pt or PtOx clusters and, in agreement with the advanced PES measurements34, identify Pt2+ as highly dispersed surface species on ceria32.
Our present study identifies the stabilization of monodispersed Pt2+ ions with one particular defect site on the ceria surface—the monoatomic step edge—and excludes the stabilization of Pt2+ on the oxygen vacancies. Monodispersed Pt on ceria is observed to be effective in incorporating excess oxygen even in the unfavourable conditions of UHV experiment. Excess oxygen and Pt2+ arrange in the square-planar PtO4 moieties decorating different types of the surface steps at coverages up to one PtO4 per one step-edge Ce atom. The excess oxygen can be easily detached, indicating enhancement of the redox properties of ceria loaded with the Pt2+ ions. Adjusting the step density and the Pt load on the model CeO2(111) surface allows maximizing the coverage of Pt2+, while suppressing the nucleation of metallic Pt0 clusters. In the present experiment, we achieve surface coverage of Pt2+ 0.05 ML. A further increase of the completely monodispersed ionized Pt2+ coverage to 0.1 ML can be expected.
Step edges on ceria have been previously identified as preferred nucleation sites for supported metal clusters20,21,27,28,35,36,37. Our present study highlights the property of the step edges on ceria to provide specific structural and electronic environments for selective formation of monodispersed, thermally and chemically stable Pt2+ ions. The step edges represent intrinsic defects ubiquitously present on nanostructured ceria surfaces4,38; our results are thus applicable for the interpretation of the properties and the optimization of the Pt2+ load on large-area ceria supports3,10,11. More generally, the step edges may represent a common type of adsorption sites providing stabilization for monodispersed metal atoms and ions in any oxide-supported single-atom catalysts15,39. Our results therefore introduce important concepts of step reactivity40 and step engineering13,14 in understanding the stability, the activity and in designing new single-atom catalysts.
Methods
Experiment
The experiments were performed on surface science apparatuses in Surface Science Laboratory in Prague (STM, laboratory X-ray PES (XPS) with hν=1,487 eV (Al Kα), low-energy electron diffraction) and at the Materials Science Beamline in Trieste (PES with hν=22–1,000 eV (synchrotron), laboratory XPS with hv=1,487 eV (Al Kα) and low-energy electron diffraction).
Preparation of the ceria substrates
The ceria layers and their Pt loading were prepared using the same procedures and parameters in both laboratories, and investigated by surface science methods in situ without exposing to air. The procedures and parameters of all samples are summarized in Supplementary Table 1. The ceria layers were prepared by deposition of Ce metal (Ce wire 99.9%, Goodfellow Cambridge Ltd) from Ta or Mo crucible heated by electron bombardment on clean Cu(111) substrate (MaTecK GmbH) in a background pressure of 5 × 10−5 Pa of O2 (5.0, Linde AG). The growth rate of CeO2 was 6 ML per hour. Varying densities of 1 ML high steps on the prepared CeO2 layers were obtained by growth of CeO2 at constant substrate temperature 423 or 523 K (Method I in Supplementary Table 1) or linearly increasing substrate temperature from room temperature to 723 K (Method II in Supplementary Table 1)16. For experiments in Fig. 1, the ordered, fully oxidized layer of CeO2 (Fig. 1a,b) and the ordered reduced layer of CeO1.7 (Fig. 1d,e) were obtained by approach published in ref. 17 that yields the lowest step density. In this approach, first, fully reduced Ce2O3 layer is prepared by depositing metallic Ce on a CeO2 layer and annealing in vacuum. Subsequently, the Ce2O3 layer is exposed to a controlled dose of O2 at 5 × 10−5 Pa and annealed to obtain desired stoichiometry CeO1.7 or CeO2 (Method III in Supplementary Table 1). The CeO2 surface imaged in Fig. 1g,h was obtained by depositing 0.3 ML CeO2 on the CeO2(111) substrate as in Fig. 1a, forming small ML-high islands. This homoepitaxy of CeO2 on CeO2 yields clearly arranged samples with high step density (Method IV in Supplementary Table 1).
Characterization of the ceria substrates
The thickness of the ceria layers was determined from the attenuation of the substrate Cu 2p3/2 XPS signal measured at hν=1,487 eV. For calculations, we used inelastic mean free path of electrons in CeO2 11.2 Å. The thickness of the ceria layers was set between 20 and 40 Å or 7 and 12 ML with 1 ML corresponding to 3.1 Å, the distance between Ce(111) atomic planes of CeO2. In this range of thickness, the coverage of the Cu substrate by ceria ranges between 97 and 100 % (ref. 17). For determining the density of 1 ML high steps, we use a semi-automated procedure when the first step outlines are marked in STM images manually. Step outlines are then mapped onto a properly scaled and rotated hexagonal mesh of surface Ce atoms. The atoms that are closest to the outlines are automatically identified as step-edge atoms and their density evaluated in ML. The error in determining the density of 1 ML high steps is estimated to be ±10 % and is marked in Fig. 2a.
Preparation of the Pt deposit
Pt was deposited on the ceria layers from a Pt wire (99.99%, MaTecK GmbH) heated by electron bombardment. Pt was deposited on the sample surface at 300 K and subsequently stabilized by increasing the sample temperature to 700 K at the rate 2 K s−1. Both Pt deposition and annealing proceeded in the UHV background pressure 5 × 10−8 Pa or below. The thermal treatment supports the ionization of Pt to Pt2+.
Characterization of the Pt deposit
The amount of Pt was calculated from the deposition time after calibrating the constant evaporation rate of the Pt evaporator. The evaporation rate was determined by a Quartz Crystal Microbalance and/or in a dedicated experiment from the thickness of 4-ML-thick Pt layers on CeO2(111)/Cu(111) determined by attenuation of the substrate Cu 2p3/2 XPS signal measured at hν=1,487 eV. This dedicated experiment was used to correlate Pt evaporation rates between the two experimental apparatuses. For calculations, we used inelastic mean free path of electrons in Pt 8 Å. The fraction of Pt2+ after thermal treatment was determined by fitting the ionic Pt2+ and neutral Pt0 component in the PES Pt 4f spectrum measured at hν=180 eV (cf. Fig. 1c,f,i). The error in determining the Pt and Pt2+ amounts on the studied samples is ±20 % and is marked in Fig. 2a. This error represents the calibration error of the Pt evaporation rate.
Resonant PES
Reduction of the ceria surface after deposition of Pt and thermal treatment was determined with resonant PES of Ce 4f state. We determine the so-called resonant enhancement ratio (RER) as defined in refs 41, 42 from measurements of intensities of Ce3+ and Ce4+ components of valence-band resonant PES Ce 4f spectra of CeO2 measured off-resonance (hν=115 eV) and on-resonance (hν=121.4 eV for the Ce3+ component and hν=124.8 eV for the Ce4+ component). The value of resonant enhancement ratio represents an upper estimate of the concentration of Ce3+ ions on the ceria surface and is plotted in Fig. 2b17,42.
STM imaging
STM measurements were performed with commercial Pt–Ir tips (Unisoku). STM imaging of CeO2(111) and Pt/CeO2(111) films was available only via unoccupied states. We used sample voltages 2.5–3.5 V and tunnelling currents 25–75 pA.
Theory
All calculations were based on the DFT and were performed using the spin-polarized GGA+U approach43, employing the Perdew–Burke–Ernzerhof exchange-correlation functional44 and ultrasoft pseudopotentials45. The spin-polarized Kohn–Sham equations were solved with a plane-wave basis set and the Fourier representation of the charge density was limited by kinetic cutoffs of 40 and 320 Ry, respectively. The Quantum-ESPRESSO computer package was used in all the calculations46. In the Hubbard U term, the occupations of the f-orbitals were defined in terms of atomic wave function projectors and the value of the parameter U was set to 4.5 eV, following our previous studies47,48.
Slab models
The ceria (111) surfaces were modelled with periodic (3 × 3) slabs being three CeO2 ML thick and separated by more than 10 Å of vacuum in the direction perpendicular to the surface. The Brillouin zone was sampled at Gamma point. In the present work, we considered two low-energy ML-high steps that we label following ref. 25 as step I and step II. The edge of both step I and step II steps are oriented along the  direction. These surface steps were modelled with vicinal surfaces described with monoclinic periodic slabs separated by >10 Å of vacuum in the direction perpendicular to the (111) terrace. The dimensions of the cells were 17.97 × 11.67 Å2 along the
direction. These surface steps were modelled with vicinal surfaces described with monoclinic periodic slabs separated by >10 Å of vacuum in the direction perpendicular to the (111) terrace. The dimensions of the cells were 17.97 × 11.67 Å2 along the  and
and  directions (step I) and 15.72 × 11.67 Å2 along the
directions (step I) and 15.72 × 11.67 Å2 along the  and
and  (step II). All the vicinal surfaces slabs included three CeO2 ML. This thickness was shown to be sufficient to calculate the structural and thermodynamic properties of these steps25. The complete set of surface structures and systems considered in this work is listed in the Supplementary Note 1. All these systems were structurally optimized according to the Hellmann–Feynman forces. During the geometry optimization, the atomic positions of the lowermost CeO2 ML were constrained, as well as those of the Ce atoms in the central ML, except for the Ce atoms below the step edge.
(step II). All the vicinal surfaces slabs included three CeO2 ML. This thickness was shown to be sufficient to calculate the structural and thermodynamic properties of these steps25. The complete set of surface structures and systems considered in this work is listed in the Supplementary Note 1. All these systems were structurally optimized according to the Hellmann–Feynman forces. During the geometry optimization, the atomic positions of the lowermost CeO2 ML were constrained, as well as those of the Ce atoms in the central ML, except for the Ce atoms below the step edge.
Energetics
Binding energies were computed as 1/NPt (Eslab+NPt EPt−Eslab/Pt), where Eslab/Pt is the total energy of the ceria slab containing NPt atoms of Pt, Eslab is the total energy of the corresponding relevant (stoichiometric, reduced or oxidized) Pt-free ceria slab and EPt is the total energy of a Pt atom in vacuum. The energies required to form an oxygen vacancy Ov were calculated as (Eslab/Ov+½ EO2−Eslab), where Eslab/Ov and Eslab are the total energies of the ceria supercell with and without the O vacancy, respectively, whereas EO2 is the total energy of a gas-phase O2 molecule compensated for the known overbinding predicted by (semi)local functionals for O2.
Additional information
How to cite this article: Dvořák, F. et al. Creating single-atom Pt-ceria catalysts by surface step decoration. Nat. Commun. 7:10801 doi: 10.1038/ncomms10801 (2016).
References
Yang, X. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Flytzani-Stephanopoulos, M. & Gates, B. C. Atomically dispersed supported metal catalysts. Annu. Rev. Chem. Biomol. Eng. 3, 545–574 (2012).
Fu, Q., Saltsburg, H. & Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 301, 935–938 (2003).
Bruix, A. et al. Maximum noble-metal efficiency in catalytic materials: atomically dispersed surface platinum. Angew. Chem. Int. Ed. 53, 10525–10530 (2014).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx . Nat. Chem. 3, 634–641 (2011).
Novotný, Z. et al. Ordered array of single adatoms with remarkable thermal stability: Au/Fe3O4(001). Phys. Rev. Lett. 108, 216103 (2012).
Parkinson, G. S. et al. Carbon monoxide-induced adatom sintering in a Pd-Fe3O4 model catalyst. Nat. Mater. 12, 724–728 (2013).
Bliem, R. et al. Subsurface cation vacancy stabilization of the magnetite (001) surface. Science 346, 1215–1218 (2014).
Li, F., Li, Y., Zeng, X. C. & Chen, Z. Exploration of high-performance single-atom catalysts on support M1/FeOx for CO oxidation via computational study. ACS Catal. 5, 544–552 (2015).
Hatanaka, M. et al. Ideal Pt loading for a Pt/CeO2-based catalyst stabilized by a Pt–O–Ce bond. Appl. Catal. B Environ. 99, 336–342 (2010).
Fiala, R. et al. Proton exchange membrane fuel cell made of magnetron sputtered Pt–CeOx and Pt–Co thin film catalysts. J. Power Sources 273, 105–109 (2015).
Gong, X.-Q., Selloni, A., Batzill, M. & Diebold, U. Steps on anatase TiO2(101). Nat. Mater. 5, 665–670 (2006).
Barth, J. V, Costantini, G. & Kern, K. Engineering atomic and molecular nanostructures at surfaces. Nature 437, 671–679 (2005).
Vang, R. T. et al. Controlling the catalytic bond-breaking selectivity of Ni surfaces by step blocking. Nat. Mater. 4, 160–162 (2005).
Gong, X., Selloni, A., Dulub, O., Jacobson, P. & Diebold, U. Small Au and Pt clusters at the anatase TiO2(101) surface: behavior at terraces, steps, and surface oxygen vacancies. J. Am. Chem. Soc. 130, 370–381 (2008).
Dvořak, F. et al. Adjusting morphology and surface reduction of CeO2(111) thin films on Cu(111). J. Phys. Chem. C 115, 7496–7503 (2011).
Duchoň, T. et al. Ordered phases of reduced ceria as epitaxial films on Cu(111). J. Phys. Chem. C 118, 357–365 (2014).
Zhou, Y., Perket, J. M. & Zhou, J. Growth of Pt nanoparticles on reducible CeO2 (111) thin films: effect of nanostructures and redox properties of ceria. J. Phys. Chem. C 114, 11853–11860 (2010).
Shao, X., Jerratsch, J.-F., Nilius, N. & Freund, H.-J. Probing the 4f states of ceria by tunneling spectroscopy. Phys. Chem. Chem. Phys. 13, 12646–12651 (2011).
Lu, J.-L., Gao, H.-J., Shaikhutdinov, S. & Freund, H.-J. Morphology and defect structure of the CeO2(111) films grown on Ru(0001) as studied by scanning tunneling microscopy. Surf. Sci. 600, 5004–5010 (2006).
Zhou, Y. & Zhou, J. Interactions of Ni nanoparticles with reducible CeO2(111) thin films. J. Phys. Chem. C 116, 9544–9549 (2012).
Mullins, D. R. et al. Water dissociation on CeO2(100) and CeO2(111) thin films. J. Phys. Chem. C 116, 19419–19428 (2012).
Bruix, A. et al. A new type of strong metal-support interaction and the production of H2 through the transformation of water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) catalysts. J. Am. Chem. Soc. 134, 8968–8974 (2012).
Negreiros, F. R. & Fabris, S. Role of cluster morphology in the dynamics and reactivity of subnanometer Pt clusters supported on ceria surfaces. J. Phys. Chem. C 118, 21014–21020 (2014).
Kozlov, S. M., Viñes, F., Nilius, N., Shaikhutdinov, S. & Neyman, K. M. Absolute surface step energies: accurate theoretical methods applied to ceria nanoislands. J. Phys. Chem. Lett. 3, 1956–1961 (2012).
Torbrügge, S., Cranney, M. & Reichling, M. Morphology of step structures on CeO2(111). Appl. Phys. Lett. 93, 073112 (2008).
James, T. E., Hemmingson, S. L. & Campbell, C. T. Energy of supported metal catalysts: from single atoms to large metal nanoparticles. ACS Catal. 5, 5673–5678 (2015).
James, T. E., Hemmingson, S. L., Ito, T. & Campbell, C. T. Energetics of Cu adsorption and adhesion onto reduced CeO2(111) surfaces by calorimetry. J. Phys. Chem. C 119, 17209–17217 (2015).
Colussi, S. et al. Nanofaceted Pd-O sites in Pd-Ce surface superstructures: Enhanced activity in catalytic combustion of methane. Angew. Chem. Int. Ed. 48, 8481–8484 (2009).
Kozlov, S. M. & Neyman, K. M. O vacancies on steps on the CeO2(111) surface. Phys. Chem. Chem. Phys. 16, 7823–7829 (2014).
Fiala, R. et al. Pt–CeOx thin film catalysts for PEMFC. Catal. Today 240, 236–241 (2015).
Hatanaka, M. et al. Reversible changes in the Pt oxidation state and nanostructure on a ceria-based supported Pt. J. Catal. 266, 182–190 (2009).
Nagai, Y. et al. Sintering inhibition mechanism of platinum supported on ceria-based oxide and Pt-oxide-support interaction. J. Catal. 242, 103–109 (2006).
Matolín, V. et al. Platinum-doped CeO2 thin film catalysts prepared by magnetron sputtering. Langmuir 26, 12824–12831 (2010).
Zhou, J., Baddorf, A. P., Mullins, D. R. & Overbury, S. H. Growth and characterization of Rh and Pd nanoparticles on oxidized and reduced CeOx(111) thin films by scanning tunneling microscopy. J. Phys. Chem. C 112, 9336–9345 (2008).
Zhou, Y., Perket, J. M. & Zhou, J. Growth of Pt nanoparticles on reducible CeO2(111) thin films: effect of nanostructures and redox properties of ceria. J. Phys. Chem. C 114, 11853–11860 (2010).
Zhou, Y. & Zhou, J. Growth and sintering of Au−Pt nanoparticles on oxidized and reduced CeOx(111) thin films by scanning tunneling microscopy. J. Phys. Chem. Lett. 1, 609–615 (2010).
Sayle, T. X. T., Parker, S. C. & Sayle, D. C. Oxidising CO to CO2 using ceria nanoparticles. Phys. Chem. Chem. Phys. 7, 2936–2941 (2005).
Castellani, N. J., Branda, M. M., Neyman, K. M. & Illas, F. Density functional theory study of the adsorption of Au atom on cerium oxide: effect of low-coordinated surface sites. J. Phys. Chem. C 113, 4948–4954 (2009).
Zambelli, T., Wintterlin, J., Trost, J. & Ertl, G. Identification of the ‘active sites’ of a surface-catalyzed reaction. Science 273, 1688–1690 (1996).
Matolín, V. et al. Water interaction with CeO2(111)/Cu(111) model catalyst surface. Catal. Today 181, 124–132 (2012).
Mullins, D. R. The surface chemistry of cerium oxide. Surf. Sci. Rep. 70, 42–85 (2015).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 71, 035105 (2005).
Perdew, J. P. J., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Fabris, S., de Gironcoli, S., Baroni, S., Vicario, G. & Balducci, G. Taming multiple valency with density functionals: a case study of defective ceria. Phys. Rev. B 71, 041102 (2005).
Fabris, S., Vicario, G., Balducci, G., De Gironcoli, S. & Baroni, S. Electronic and atomistic structures of clean and reduced ceria surfaces. J. Phys. Chem. B 109, 22860–22867 (2005).
Acknowledgements
This work was supported by Czech Science Foundation (contract numbers 15-06759S and 13-10396S), and by the European Union via the FP7-NMP-2012 project chipCAT under contract number 310191 and the EU FP7 COST action CM1104. A.T. acknowledges the support of the Grant Agency of the Charles University, contract number 2048514. S.F. acknowledges the support provided by the Humboldt Foundation through a Friedrich Wilhelm Bessel Research Award. The high-performance computing resources were gratefully provided by ISCRA initiative of CINECA. CERIC-ERIC consortium is acknowledged for financial support.
Author information
Authors and Affiliations
Contributions
F.D., A.T., M.V., T.S., I.M., J.M. and V.M. designed and performed the experiments. M.F.C., N.-D.T., F.R.N. and S.F. performed the DFT calculations. All authors interpreted the experimental and computational results. J.M., S.F., T.S., I.M. and V.M. wrote the manuscript. F.D., A.T., J.M., I.M., V.M. and S.F. provided funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-7, Supplementary Tables 1-2, Supplementary Note 1, Supplementary Discussion and Supplementary References (PDF 2431 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dvořák, F., Farnesi Camellone, M., Tovt, A. et al. Creating single-atom Pt-ceria catalysts by surface step decoration. Nat Commun 7, 10801 (2016). https://doi.org/10.1038/ncomms10801
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms10801
This article is cited by
-
The reformation of catalyst: From a trial-and-error synthesis to rational design
Nano Research (2024)
-
Memory-dictated dynamics of single-atom Pt on CeO2 for CO oxidation
Nature Communications (2023)
-
Grain boundary boosting the thermal stability of Pt/CeO2 thin films
Nano Research (2023)
-
Recent advances in regulating the local environment of M-N4 structure for tailored chemical reactions
Nano Research (2023)
-
Data-driven models for ground and excited states for Single Atoms on Ceria
npj Computational Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
