Abstract
The lysosomal–autophagic pathway is activated by starvation and plays an important role in both cellular clearance and lipid catabolism. However, the transcriptional regulation of this pathway in response to metabolic cues is uncharacterized. Here we show that the transcription factor EB (TFEB), a master regulator of lysosomal biogenesis and autophagy, is induced by starvation through an autoregulatory feedback loop and exerts a global transcriptional control on lipid catabolism via Ppargc1α and Ppar1α. Thus, during starvation a transcriptional mechanism links the autophagic pathway to cellular energy metabolism. The conservation of this mechanism in Caenorhabditis elegans suggests a fundamental role for TFEB in the evolution of the adaptive response to food deprivation. Viral delivery of TFEB to the liver prevented weight gain and metabolic syndrome in both diet-induced and genetic mouse models of obesity, suggesting a new therapeutic strategy for disorders of lipid metabolism.
Similar content being viewed by others
Main
The adaptive response of an organism to food deprivation is associated with major transcriptional and metabolic1,2,3,4,5 changes and is conserved across evolution6,7. One of the most prominent metabolic changes observed during starvation is an increase in lipid catabolism in the liver.
Autophagy, a lysosome-dependent catabolic process, is activated by starvation8 and the resulting breakdown products are used to generate new cellular components and energy. Recent studies revealed that autophagy plays a central role in lipid metabolism because it shuttles lipid droplets to the lysosome where they are hydrolysed into free fatty acids (FFAs) and glycerol9,10. Moreover, excessive lipid overload may inhibit autophagy, whereas enhancing liver autophagy in murine genetic models of obesity (Ob/Ob) ameliorates their metabolic phenotype11. These observations indicate the close relationship between intracellular lipid metabolism and the lysosomal–autophagic pathway. However, it is not clear how this relationship is coordinated at the transcriptional level in response to environmental cues. Here we show that the basic helix–loop–helix (bHLH) leucine zipper transcription factor TFEB, a master regulator of lysosomal biogenesis and autophagy12,13, mediates the organismal transcriptional and metabolic responses to starvation.
Starvation induces Tcfeb expression through an autoregulatory loop
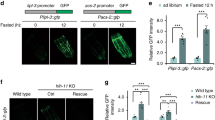
We investigated whether starvation activated Tcfeb transcription. Tcfeb expression was significantly induced in liver, muscle and kidney of mice subjected to 24 h of food deprivation (Fig. 1a). Similar results were observed in vivo using a heterozygous transgenic mouse line (Tcfeb– β-gal) that carries a Tcfeb gene-trap allele fused with the β-galactosidase gene. In this model we found stronger β-galactosidase staining in the liver and kidney of fasted mice when compared with fed mice (Fig. 1b). Starvation time-course studies of HeLa cells, mouse embryonic fibroblasts (MEFs) and hepatocytes revealed a significant and progressive increase of TFEB messenger RNA and protein expression levels starting as soon as 4 h after the elimination of nutrients from the culture medium (Fig. 1c and Supplementary Fig. S1a). Notably, the cytoplasm-to-nucleus translocation of TFEB (ref. 13) occurs at an earlier time point after starvation when compared with TFEB transcriptional activation (Supplementary Fig. S1b). These observations led us to predict that TFEB exerted a positive effect on its own transcription.

(a) Expression levels of Tcfeb mRNA in tissues isolated from 24-h-fasted (24 h starved) 6-week-old mice. Values are expressed as fold change relative to Tcfeb expression in mice fed ad libitum (Fed). Bars represent mean ± s.d. for n = 5 mice; *P≤0.05; **P≤0.01; ***P≤0.001. (b) Representative β-gal staining of liver and kidney frozen sections isolated from fed and 24-h-fasted heterozygous Tcfeb– β-gal mice. (c) Time-course expression analysis of TFEB in wild-type HeLa cells, MEFs and hepatocytes after the addition of starvation media (time 0). Data represent mean ± s.d. for n = 3 independent experiments. (d) Expression levels of transfected hTFEB–FLAG, Tcfeb– β-gal fusion transcript and endogenous Tcfeb mRNAs in MEFs isolated from control (+/+) and heterozygous Tcfeb– β-gal/+ mice. Data represent mean ± s.d. for n = 3 mice; *P≤0.05. (e) Time-course expression analysis of TFEB mRNA in control (Ctrl) or TFEB-overexpressing HeLa cells during fasting and re-feeding. Primers specific for the endogenous TFEB were used. Data represent mean ± s.d. for n = 3. (f) Time-course expression analysis of Tcfeb mRNA in heterozygous Tcfeb– β-gal/+ or control MEFs during fasting and re-feeding. Specific primers for the endogenous Tcfeb were used. Data represent mean ± s.d. for n = 3 mice. (g) Expression analysis of Tcfeb mRNA in heterozygous Tcfeb– β-gal/+ or control hepatocytes in fed and after 24 h fasting. Data represent mean ± s.d. for n = 3 mice. (h) ChIP analysis from liver of mice fed ad libitum or 24 h fasted. The CLEAR elements in the first intron of Tcfeb genomic DNA are shown as numbered black rectangles as indicated in Supplementary Table S1. Red rectangles represent exons, and the ATG indicates the first codon (from the mouse Tcfeb isoform b). The histogram shows the amount of immunoprecipitated DNA as detected by quantitative PCR assay. Values were normalized to the input and plotted as relative enrichment over a mock control. Data represent mean ± s.d of 3 independent experiments.
The overexpression of the human TFEB complementary DNA in MEFs from heterozygous Tcfeb– β-gal transgenic mice resulted in a significant increase in the transcription of the Tcfeb– β-galactosidase fusion transcript, indicating that the endogenous Tcfeb gene is positively regulated by the exogenous TFEB (Fig. 1d). This result was also confirmed using a set of primers that specifically amplifies the murine endogenous Tcfeb transcript, but does not amplify the exogenous human TFEB cDNA (Fig. 1d). These results indicate that exogenous TFEB can induce endogenous Tcfeb expression and suggest the presence of a positive feedback loop.
The positive feedback of TFEB on its own expression was significantly enhanced during starvation and was suppressed by re-adding nutrients to the culture medium (Fig. 1e). In addition, the effect of starvation on Tcfeb expression was significantly reduced in MEFs and hepatocytes from heterozygous mice carrying a Tcfeb– β-gal allele that lacks the transcriptional transactivation domain, indicating that the positive feedback loop requires a functional Tcfeb allele and that Tcfeb induction by starvation is sensitive to Tcfeb copy number (Fig. 1f,g).
Next, we investigated whether Tcfeb mediates the positive feedback loop on its own expression by directly binding to its promoter. We previously showed that TFEB recognizes E-box-type DNA sequences, named CLEAR motifs14. Sequence analysis identified 6 putative CLEAR sites in the promoter region of the Tcfeb gene (Fig. 1h and Supplementary Table S1). Chromatin immunoprecipitation coupled with quantitative PCR (ChIP–qPCR) of liver samples from starved transgenic mice overexpressing a tagged version of Tcfeb (ref. 13; Supplementary Fig. S2a) showed an enhancement of Tcfeb binding to CLEAR sites 1, 3, 5 and 6 when compared with fed controls (Fig. 1h). Thus, in vitro and in vivo data showed that TFEB controls its own expression by virtue of starvation-induced direct binding to CLEAR elements in the TFEB promoter.
TFEB regulates genes involved in lipid metabolism through Ppargc1α and Ppar1α
To determine whether TFEB is involved in the metabolic response to starvation we sought to define the complete TFEB-dependent transcriptome in the liver, a primary site for the organismal starvation response. To this end, we injected mice with an adenoviral vector that expresses human TFEB (HDAd-TFEB) under the control of a liver-specific promoter (PEPCK) and with a transgeneless control vector. The levels of expression of TFEB protein under these conditions are shown in Supplementary Fig. S2b. Microarray analysis indicated that as a result of TFEB overexpression, 773 genes were upregulated and 611 genes were downregulated (GSE35015), using a threshold for statistical significance (false discovery rate (FDR)<0.05) and further filtering with an absolute fold change ≥1.5. Microarray results were validated by quantitative real-time PCR (rtPCR) performed on 40 selected genes (Fig. 2a,b). Surprisingly, the gene ontology category15,16 most significantly upregulated by TFEB overexpression was the cellular lipid metabolic process, which includes monocarboxylic acid, fatty acid and cellular ketone metabolic processes, among others (Supplementary Table S2). Interestingly, several gene categories related to lipid biosynthesis, such as steroid, lipid and isoprenoid biosynthetic processes, were significantly downregulated (Supplementary Table S3). We also observed that in liver, TFEB positively regulates the expression of several genes involved in lysosome organization and autophagy (Supplementary Table S2 and Fig. 2b), which is consistent with previous results obtained in cultured cell lines12,13,14.

(a,b) mRNA levels of the indicated genes involved in lipid metabolism (a) and in the autophagy and lysosome pathway (b) were quantified by quantitative rtPCR of total RNA isolated from liver samples of mice infected with the HDAd-TFEB virus. Gapdh was used as a control (Ctrl). Values are mean ± s.d for n = 3 mice and are expressed as fold increase compared with control mice (injected with transgeneless viral vector). *P<0.05; **P<0.01; ***P<0.001. Control levels are indicated by dashed lines. (c) The 124 genes with a known role in the lipid metabolic process, whose expression was perturbed by TFEB overexpression, are represented as coloured circles and assigned to specific lipid breakdown (left) or lipid biosynthesis (right) sub-categories. The percentages of upregulated (red circles) and downregulated genes (green circles) are shown both for the two main groups and for each sub-category. *Note that in calculating these percentages, genes assigned to the negative regulation of lipid catabolic process and to the negative regulation of lipid biosynthetic process have been included in the lipid breakdown and in the lipid biosynthesis groups, respectively.
Overall we found that the transcriptional signature of TFEB overexpression in liver was similar to that of starvation (GSE36510; Supplementary Table S4), particularly for genes involved in lipid metabolism (Supplementary Fig. S3 and Tables S5 and S6). The global control of lipid metabolism exerted by TFEB is illustrated in Fig. 2c and Supplementary Table S7. These results strongly suggested that TFEB overexpression in fed mice phenocopied the transcriptional effects of nutrient deprivation in vivo, supporting the concept that TFEB is a critical regulator of the response to starvation in the liver.
Interestingly, one of the genes whose expression was significantly upregulated following TFEB overexpression was the peroxisome proliferator-activated receptor γ coactivator 1 α (Ppargc1a; also known as Pgc-1α) a known key regulator of liver lipid metabolism that is transcriptionally induced during starvation 17,18. To determine whether Pgc-1α is a direct target of Tcfeb we analysed its promoter and identified three CLEAR sites. ChIP–qPCR from liver extracts showed that Tcfeb binds to two of these sites in a starvation-dependent manner (Fig. 3a). Furthermore, transactivation of a Pgc-1α promoter luciferase reporter by TFEB was dependent on the CLEAR sites (Fig. 3b,c) and was enhanced by starvation (Fig. 3d). These results indicated that Tfeb directly regulates Pgc-1α gene expression.

(a) ChIP analysis from liver of mice fed ad libitum (fed) or 24 h-fasted (starved). CLEAR sites in the promoter region of Pgc-1α are indicated by squares. Numbers indicate the distance (bp) of the binding element from the start codon. Bar graphs show the amount of immunoprecipitated DNA as detected by quantitative PCR assay. Values were normalized to the input and plotted as relative enrichment over a mock control. Bar graphs represent mean ± s.d of 3 independent experiments, *P≤0.05; **P≤0.01. (b) Representative diagrams of the constructs containing the promoter region of Pgc-1α with either intact (Pgc-1α WT) or deleted (Pgc-1α DEL) CLEAR elements upstream of the luciferase cDNA. (c) Luciferase activity was measured after transfecting increasing amounts of TFEB–FLAG in combination with Pgc-1α WT or Pgc-1α DEL plasmids. Bar graphs represent mean ± s.d. of n = 3independent experiments, ****P≤0.0001 compared with mock transfected cells. (d) Luciferase activity was measured in cells stably overexpressing TFEB cultured in normal and starved media. Bar graphs represent mean ± s.d. of n = 3independent experiments **P≤0.01 compared with mock transfected cells. (e) Quantification of mRNA levels of PGC1α in liver and hepatocytes from control, HDAd-TFEB and Tcfeb-LiKO (liver KO) mice treated as indicated. Bar graphs show mean ± s.d. for n = 4 mice. *P≤0.05; **P≤0.01. (f) Expression analysis of TFEB target genes in liver of mice with indicated genotypes. Bar graphs show mean ± s.d. for n = 3 mice. *P≤0.05;**P≤0.01; ***P≤0.001. (g) Expression analysis of PGC1α/PPARα target genes in liver from either fasted or fed mice with indicated genotypes. Bar graphs show mean ± s.d. for n = 4. *P≤0.05; **P≤0.01 compared with the respective controls (fed or fasted).
These data were confirmed in vivo by measuring and comparing Pgc-1α expression in mice that overexpress TFEB in the liver and in mice that lack Tcfeb in the liver (Tcfeb-LiKO) with corresponding control mice. Injection of HDAd-TFEB into the liver of fed animals was sufficient to drive high constitutive Pgc-1α expression to levels similar to those observed in uninjected starved animals. In contrast, Pgc-1α induction by starvation was partially blocked by the deletion of Tcfeb. Furthermore, primary hepatocytes from animals overexpressing TFEB showed high constitutive expression of Pgc-1α, whereas Tcfeb deletion did not affect basal Pgc-1α expression. Conversely, Pgc-1α induction was partially blunted in starved hepatocytes lacking Tfeb, indicating that starvation induces Pgc-1α in a TFEB-dependent manner. In starved hepatocytes, overexpression of TFEB caused synergistic induction of Pgc-1α expression beyond the levels obtained by starvation alone (Fig. 3e). Together, these results strongly suggested that TFEB directly controls Pgc-1α induction during liver starvation response, and that the level of TFEB expression is a critical parameter for the magnitude of this response.
To study the role of Pgc-1α as a mediator of TFEB function, we overexpressed TFEB in a mouse line that lacks Pgc-1α in the liver, which was generated by injecting Pgc -1α flox/flox mice with a helper-dependent adenovirus containing CRE recombinase (HdAD-APOA1-CRE). Figure 3f shows that TFEB-mediated induction of the expression of genes involved in lipid metabolism was severely reduced in the absence of Pgc-1α, confirming that Pgc-1α acts downstream of TFEB and mediates TFEB function.
During starvation, PGC1α regulates lipid metabolism in the liver through the downstream nuclear receptor peroxisome proliferator activated receptor α (PPARα; refs 17, 18), suggesting that PGC -1α may mediate TFEB function by controlling the activity of PPARα. To test this hypothesis, we analysed transcript levels for known targets of PPARα (refs 17, 18, 19, 20) in Tcfeb-LiKO mice, compared with wild-type mice. Starvation caused induction of the Pparα target genes in wild-type mice, but not in Tcfeb-LiKO mice (Fig. 3g and Supplementary Fig. S4a), indicating that TFEB is essential for Pparα activation by starvation. Furthermore, most (74%) of the genes that were induced by TFEB overexpression in the liver of wild-type mice failed to show a transcriptional induction in mice lacking Pparα as measured by microarray analysis of liver tissue (GSE41141; Supplementary Table S8), suggesting that PPARα is an important mediator of TFEB transcriptional activity in the liver during starvation.
TFEB regulates lipid breakdown in the liver
Histological analysis of liver samples did not reveal any significant differences between wild-type and Tcfeb-LiKO mice fed with a normal diet. However, after a 24 h fast we observed an accumulation of lipid droplets in Tcfeb-LiKO mice that was not found in wild-type littermates, suggesting a defect in intracellular lipid degradation (Fig. 4a–c). Consistently, we detected an impairment of FFA oxidation in cultured hepatocytes (Fig. 4d) and higher levels of circulating FFA (Fig. 4e) and glycerol (Fig. 4f) in Tcfeb-LiKO mice when compared with controls. In addition, during starvation Tcfeb-LiKO mice showed decreased plasma levels of circulating ketone bodies, which are mainly produced in the liver from the oxidation of fatty acids (Fig. 4g). These data demonstrate the importance of TFEB in the control of cellular lipid metabolism. In addition, EchoMRI analysis of whole-body compositions demonstrated a defective peripheral fat mobilization after a 24 h and a 48 h fast (Fig. 4h). This observation could explain the increased peripheral adiposity in Tcfeb-LiKO mice when compared with controls (Fig. 4i), indicating that TFEB activity in liver also affects peripheral fat metabolism.

(a) Oil red O staining of liver sections isolated from mice with the indicated genotype fed ad libitum and 24 h fasted. Original magnification ×40. (b) Toluidin blue staining of liver sections isolated from fed and 24-h-fasted Tcfeb-LiKO and control mice (Tcfeb flox/flox mice). Arrows indicate lipid droplets. Original magnification ×100. (c) Bar graphs show the quantification of the number of lipid droplets per hepatocyte from electron microscopy analysis. A representative image is shown on the right. Values are mean ± s.d. of at least 10 cells per mouse (n = 3 mice per group). *P≤0.05. (d) The oxygen consumption rate (OCR) in primary hepatocytes isolated from control and Tcfeb-LiKO mice was measured with an XF24 analyser (Seahorse) before and after the addition of palmitic acid (0.2 mM) conjugated with BSA. The vertical red line indicates the time at which palmitate was added to cells. Values are mean ± s.d. for 3 independent experiments *P≤0.05. (e,f) Total FFA (e) and glycerol (f) in the serum isolated from 6-h-fasted Tcfeb-LiKO and control mice. Values are mean ± s.d. (n = 5 mice per group) *P≤0.05 compared with controls. (g) Total serum ketones in fed and fasted Tcfeb-LiKO and control mice. Bars are mean ± s.d. for n = 10 mice per group. **P≤0.01 compared with fed control mice. (h) EchoMRI measurement of fat and lean mass in fed and in 24- and 48-h-fasted mice expressed as relative percentage to fed (100% in the graph). Indicated values are mean ± s.d. for n = 5 mice per group. *P≤0.05;**P≤0.01 compared with fed control mice. (i) Visceral fat pad mass isolated from 2-month-old-mice with indicated genotypes. Indicated values are mean ± s.d. for n = 5 mice per group. *P≤0.05; compared with fed control mice.
Next we addressed the role of TFEB in fat storage and its utilization in animals challenged with a high-fat diet. Liver appearance and lipid content were examined in Tcfeb-LiKO, HDAd-TFEB-injected and in corresponding control mice. Livers from Tcfeb-LiKO mice were large, pale and filled with lipid vacuoles, consistent with an impairment of lipid degradation pathways. Control mice fed with the same diet showed a similar, albeit milder, phenotype, in spite of similar food intake. Conversely, livers from HDAd-TFEB-injected mice exhibited normal red colour, markedly reduced lipid content when compared with wild-type controls, and normal weight in spite of increased food intake when compared with control mice, suggesting that TFEB overexpression prevented the effects of the high-fat diet by enhancing lipid degradation (Fig. 5a–c and Supplementary Fig. S4b).

(a) Mice with the indicated genotype were kept on a high-fat diet (HFD) for 12 weeks when indicated. Gross liver morphology (upper panel), haematoxylin–eosin (H/E) (middle panel), and oil red O staining of liver sections (bottom panel). Ctrl, control. (b,c) Bar graph showing normalized liver weights (mean ± s.d. for n = 10 mice per group; b) and total lipid content in mice with indicated genotype (mean ± s.d. for n = 10; c). *P≤0.05;***P≤0.001 compared with control. (d) Toluidin blue staining of liver sections isolated from Atg7-LiKO mice injected with HDAd-Ctrl or HDAd-TFEB vector. (e,f) Bar graph showing normalized liver weights (mean ± s.d. for n = 10 mice per group ***P≤0.001; e) and total lipid content in mice with the indicated genotype (mean ± s.d. for n = 5 mice per group; ***P≤0.001; NS, not significant; f). Mice injected with an empty HDAd virus behaved as wild-type untreated mice; therefore, data are not represented in the figure.
Blocking autophagy in the liver leads to hepatomegaly and liver failure21. Recently, a significant increase of lipid droplets, cholesterol and triglycerides was observed in the liver of Atg7 KO mice, suggesting a role for autophagy in lipid degradation9. We analysed TFEB-mediated lipid degradation in Atg7 liver-KO mice, in which autophagy is blocked21. Liver-specific Atg7 KO mice were generated by injecting Atg7 flox/flox mice with a helper-dependent virus containing CRE recombinase (HdAD-APOA1-CRE; Supplementary Fig. S5a). One month after TFEB injection, the mice presented a significant increase in liver size and increased markers of liver damage, as measured by alanine transaminase, aspartate aminotransferase and alkaline phosphatase, which is consistent with previous results21. At the cellular level, we observed an accumulation of P62 (ref. 22; Supplementary Fig. S5b) and lipid droplets (Fig. 5d–f). However, TFEB overexpression failed to decrease lipid droplet number (Fig. 5d), liver weight gain (Fig. 5e) and lipid content (Fig. 5f) in Atg7 liver-KO mice (Supplementary Fig. 5b,c), which is in contrast to the results obtained by TFEB overexpression in wild-type mice (Fig. 5a–c). These results indicate that autophagy is required for TFEB-mediated lipid degradation.
TFEB overexpression rescues obesity and metabolic syndrome in mice
When fed a regular chow diet, HDAd-TFEB-injected mice were significantly leaner than controls (Fig. 6a), with decreased body fat deposition (Fig. 6b), in spite of exhibiting similar food intake. Interestingly, these differences were blunted when HDAd-TFEB was injected into Pparα KO mice (Fig. 6a,b), consistent with our results showing that Pparα is required for transcriptional changes downstream of TFEB. HDAd-TFEB-injected wild-type mice also exhibited lower plasma levels of total cholesterol, triglycerides, very-low-density lipoprotein, leptin, insulin and glucose (Fig. 6c–i). Indirect calorimetric analysis revealed a decrease in respiratory exchange rate (Fig. 6j) and an increase in FFA oxidation rate in HDAd-TFEB-injected mice when compared with controls (Fig. 6k). Overall, the phenotype observed in HDAd-TFEB-injected mice shared some similarities to those reported for mice under caloric restriction23.

(a,b) Body weight (a) and visceral fat mass (b) isolated from 2-month-old-mice with the indicated genotypes. n = 5 mice per group. (c–i) Serum metabolic profile in HDad-TFEB mice compared with control mice. (j,k) Respiratory exchange ratio (RER; ; j) and fatty acid utilization calculated from data in j (k). Values are mean ± s.d (n = 10 mice per group) *P≤0.05;**P≤0.01; compared with controls. VLDL, very-low-density lipoprotein; HDL, high-density lipoprotein.
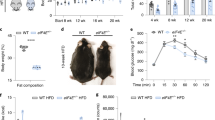
Next we investigated the metabolic effects of TFEB gain and loss of function in mice fed with a high-fat diet. We found that in this condition Tcfeb-LiKO mice gained substantially more weight than control littermates (Fig. 7a). Conversely, TFEB overexpression by injection of HDAd-TFEB at the beginning of the high-fat diet (Early inj) significantly prevented the development of obesity. Furthermore, the injection of HDAd-TFEB after 4 weeks of high-fat diet (Late inj) completely arrested the development of the obese phenotype. In addition, after 10 weeks of high-fat diet the weights of the animals injected with HDAd-TFEB at the beginning of the experiment and those injected after 4 weeks were indistinguishable (Fig. 7a). Body composition analysis revealed that the weight difference between Tcfeb-LiKO and HDAd-TFEB-injected mice was largely due to differences in fat accumulation, which was higher in Tcfeb-LiKO and lower in HDAd-TFEB-injected mice when compared with controls (Fig. 7b). Of note, the abnormalities in the serum metabolic profile induced by the high-fat diet in wild-type mice, which was characterized by the increase of circulating leptin, insulin, triglycerides and cholesterol, were markedly attenuated in HDAd-TFEB-injected mice (Fig. 7c–f), which also showed improved sugar metabolism, as demonstrated by glucose and insulin tolerance tests (Fig. 7g–i).

(a) Body weight curves of male mice fed with the high-fat diet (HFD; 40% calories from fat) for 10 weeks starting from 5 weeks of age (0 on the x axis). Mice were injected with HDAd-TFEB either 1 week before (early inj), or 4 weeks after, (late inj) being placed on the high-fat diet, as indicated by the arrows. Values are represented as percentages of weight increase. Ctrl, control. (b) Whole-body composition analysis (EchoMRI) of the same mice as in a after 10 weeks of the high-fat diet. In a,b n = 10 mice per group; bars represent mean ± s.d. *P≤0.05;**P≤0.01; ***P≤0.001 compared with control high-fat diet group. (c–f) Total serum insulin, leptin, triglyceride and cholesterol levels in control and HDAd-TFEB mice kept on the high-fat diet for 10 weeks. Value are mean ± s.d. n = 10 add per group. *P≤0.05; ****P≤0.0001 compared with control. (g–i) Glucose and insulin tolerance tests in control and HDAd-TFEB mice challenged with the high-fat diet for 10 weeks. (g,h) Glucose (g) and serum insulin (h) levels at the indicated time points after glucose challenge. (i) Glucose levels at the indicated time points after insulin challenge. In g–i, values are mean ± s.d. n = 7 mice per group; *P≤0.05;**P≤0.01 compared with control. Mice injected with an empty HDAd virus behaved as wild-type untreated mice; therefore, data are not represented in the figure.
Similar experiments were performed in a genetic model of obesity due to leptin deficiency (Ob/Ob), which is associated with hyperphagia24. Two-month-old Ob/Ob mice, already obese at this early age, were injected with HDAd-TFEB. Six weeks later, HDAd-TFEB-injected Ob/Ob mice had significantly reduced levels of circulating triglycerides, cholesterol, glucose and insulin, and an improved glucose tolerance test, compared with untreated Ob/Ob mice, indicating that liver overexpression of TFEB improved the metabolic syndrome phenotype (Supplementary Fig. S6). Together, these observations demonstrate that high TFEB activity can not only prevent the metabolic syndrome, but also revert it once initiated.
Evolutionary conservation of TFEB regulation and function in C. elegans
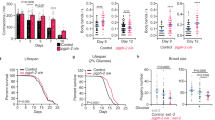
The genetically tractable nematode C. elegans shares approximately 80% of its genes with humans25. The C. elegans genome encodes a single homologue of TFEB, the gene hlh-30 (Supplementary Fig. S7). Its protein product HLH-30 was previously shown to recognize a DNA motif similar to the CLEAR motif in vitro, and to drive transcription of metabolic genes in vivo26. To assess evolutionary conservation of TFEB-mediated starvation responses, we investigated whether HLH-30 may act in a similar manner to TFEB during C. elegans starvation. First, we found that hlh-30 mRNA progressively accumulated over a time course of starvation in wild-type animals, and rapidly decreased after reintroduction of food, similar to mammalian TFEB (Fig. 8a). In contrast, the level of hlh-30 transcript did not increase after 12 h of starvation in hlh-30(tm1978)-null mutants (Fig. 8b), suggesting that hlh-30 is induced in an hlh-30- dependent manner during starvation by an autoregulatory feedback loop, similar to the mammalian TFEB. C. elegans intestinal cells perform similar metabolic functions to the vertebrate liver, including lipid storage and metabolism. Using semi-quantitative oil red O staining, we found that starved wild-type animals consumed approximately 20% of their lipid stores, compared with well-fed counterparts (Fig. 8c–g). Starved hlh-30 mutants exhibited a significantly smaller reduction in lipid staining, close to 10%, suggesting that they failed to mobilize lipids as promptly as wild-type animals. Consistently, using transmission electron microscopy, we found that intestinal cells in starved wild-type animals became depleted of dark-staining lipid droplets (Fig. 8h,i), whereas those of starved hlh-30 mutants exhibited abundant droplets (Fig. 8j,k). These results suggest that nematodes, similarly to mice, require HLH-30/TFEB to efficiently use lipid stores during periods of starvation. Furthermore, as in liver TFEB-deficient mice, lipid catabolism gene induction is greatly compromised in starved hlh-30 mutants (Fig. 8l). These data suggest that the reason starved hlh-30 mutants fail to mobilize their lipid stores may be because of a severe transcriptional response defect. In wild-type C. elegans, starvation results in lifespan extension27 (Fig. 8m). In contrast, loss of hlh-30 resulted in almost complete abrogation of starvation-induced lifespan extension (Fig. 8n). In addition, first-stage (L1) wild-type larvae arrest in response to food deprivation, resuming development following food restoration 28. In contrast, hlh-30 mutant L1 larvae completely failed to survive starvation-induced arrest, indicating that hlh-30 is not only required in adult animals to survive starvation, but in younger stages as well (Fig. 8o). Considered together, these data demonstrate how hlh-30 expression is induced, that it is required for lipid mobilization, is necessary for a proper transcriptional response, and is required for survival, all during starvation. Therefore, our observations suggest that HLH-30 and murine TFEB share evolutionarily conserved functions in organismal adaptation to starvation.

(a) hlh-30 quantitative rtPCR showing increased expression of the C. elegans TFEB gene hlh-30 over a time course of starvation in wild-type animals followed by a rapid decrease to the basal level following re-feeding of the animals (mean±s.e.m. of n = 3). *P≤0.05 (t-test, compared with wild-type at t = 0 h). (b) hlh-30 3’ UTR quantitative rtPCR showing expression of hlh-30 after 12 h starvation in wild-type and hlh-30(tm1978) animals (mean±s.e.m. of n = 3). *P≤0.05 (t-test, compared with wild-type starved). (c) Quantification of oil red O stain in starved animals relative to well-fed counterparts (mean±s.e.m. of n = 3). **P≤0.01 (t-test, compared with wild-type starved). (d–g) Representative micrographs of wild-type and hlh-30 animals after 8 h starvation and stained with oil red O. (h–k) Representative TEMs of wild-type and hlh-30 animals after 24 h starvation. IEC, intestinal epithelial cell; EPI, epidermis; BB, brush border; GON, gonad; BWM, body wall muscles, aj, apical junction; ld, lipid droplet. Scale bar, 2 μm. (l) Quantitative rtPCR of starvation-induced genes in wild-type and hlh-30 animals after 12 h starvation (means ± s.e.m of n = 3). *P≤0.05; **P≤0.01; ***P≤0.001 (t-test, compared with wild-type starved). (m,n) hlh-30 is required for starvation-induced lifespan extension. One representative experiment of 2 independent trials; error bars represent mean±s.e.m. Median survival (MS) (wild-type fed) = 10 d; MS (wild-type starved) = 17 d, P<0.0001 versus fed using the Log-rank test; MS (hlh-30 fed) = 7 d; MS (hlh-30 starved) = 9 d, P<0.0001 versus fed. (o) L1 arrest assay showing survival of wild-type and hlh-30 starved animals relative to non-starved conditions (mean±s.e.m. of n = 3). ***P≤0.001 (t-test, compared with wild-type starved).
DISCUSSION
Our study identifies TFEB as a key player in the metabolic response to starvation. TFEB activity is regulated transcriptionally and post-transcriptionally by nutrients, and is required to induce starvation-response genes in both mammals and worms. Most importantly, the absence of TFEB results in an impairment of lipid catabolism and in a more severe metabolic derangement in obese animals, whereas TFEB overexpression causes the opposite effects and rescues obesity and associated metabolic syndrome.
TFEB mRNA expression is induced by starvation by a post-transcriptional switch that controls TFEB nuclear translocation29,30,31, which allows TFEB to rapidly respond to nutrient availability, and a positive transcriptional autoregulatory component for a sustained response.
Autoregulatory feedback circuits are used by eukaryotic cells to convert a graded input into a binary (ON/OFF) response in eukaryotic gene circuits32,33,34. Interestingly, the Pgc-1α gene, a direct target of TFEB, is also subject to an autoregulatory loop35.
Notably, TFEB regulates genes involved in several steps of lipid catabolism, which occur in different cellular compartments, such as the transport of fatty acid chains across the plasma membrane (for example, Cd36 and Fabps), and the β-oxidation of FFA in mitochondria (for example, Cpt1, Crat, Acadl, Acads and Hdad) and in peroxisomes (Cyp4a genes). According to our data, most of the effects of TFEB on lipid metabolism seem to be mediated by the direct regulation exerted by TFEB on the Pgc-1α–Ppar -α complex. Interestingly, a recent study showed that Pgc-1α participates in the transcriptional co-activation of TFEB, although the magnitude of this effect seems to be modest36.
The observation that TFEB gain and loss of function in the liver influences whole-body energy metabolism suggests that TFEB stimulates the liver secretion of factors that affect the function of other tissues. This is likely to be mediated by Pgc-1α and Ppar -α, which are known to regulate the production of secreted hormones37,38,39.
In previous studies we demonstrated that TFEB controls autophagy by directly regulating lysosomal and autophagy genes12,13. Interestingly, the overexpression of TFEB in mice in which autophagy was genetically suppressed by deletion of Atg7 in the liver did not rescue hepatic steatosis, suggesting that TFEB effects on lipid metabolism require a functional autophagic pathway. Thus, TFEB controls the starvation response by orchestrating the induction of autophagy and Pgc-1α–Ppar -α -mediated lipid catabolism. We propose a model (Supplementary Fig. S8) in which adequate nutrition keeps TFEB inactive by cytoplasmic sequestration. During starvation, TFEB translocates to the nucleus where it induces its own expression. This initiates a fast and pronounced metabolic shift to the catabolism of energy stores. On nutrient restoration, the feedback loop is quickly interrupted by TFEB nuclear exclusion, restoring the system to baseline. It is likely that global control of lipid metabolism by TFEB arose early during evolution to facilitate organismal adaptation to challenging nutritional conditions, as evidenced by the evolutionary conservation of TFEB autoregulation and by its role in metabolic adaptation to starvation in the invertebrate C. elegans. Finally, the acute beneficial effects of TFEB overexpression in both diet- and genetically induced obese mice suggest that this regulatory circuit may be an attractive therapeutic target for the modulation of lipid metabolism in obesity-related diseases.
METHODS
Generation of mouse models.
Conditional Tcfeb-flox and Tcfeb–3xFLAG transgenic mouse line generation was described previously11,29. The gene-trapping Tcfeb–β-gal mouse line was obtained by injecting an embryonic stem cell clone carrying a β-galactosidase coding sequence into the third intron of the Tcfeb genomic DNA of a blastocyst. The following mice were obtained from the Jackson laboratory: Albumin–CRE (ALB–CRE), the Ob/Ob line, the Ppar KO line and the Pgc-1α f/f mouse line. The Atg7 f/f mouse line was obtained from N. Mizushima (Tokyo Medical and Dental University Graduate School and Faculty of Medicine, Japan). All mice used were males and maintained in a C57BL/6 strain background. Fasting experiments were carried out for 24 h starting from the morning (otherwise indicated). In the high-fat diet study, age-matched male mice were fed ad libitum a Western-style diet (Harlan Teklad TD 88137) containing 21% (w/w) total lipid (42% calories as anhydrous milk fat). Body weights were recorded bi-monthly. For all experiments involving Tcfeb-LIKO mice, the control mice were Tcfeb loxP/loxP mice that did not carry the ALB-CRE transgene. All experiments were approved by the Committee on Animal Care at Baylor College of Medicine and conform to the legal mandates and federal guidelines for the care and maintenance of laboratory animals.
HDAd virus production.
The HDAd-TFEB virus contains the following elements (from 5′ to 3′): a liver-restricted rat phosphoenolpyruvate carboxykinase promoter40, the ApoAI intron, the hTFEB cDNA, the woodchuck hepatitis virus post-transcriptional regulatory element, the ApoE locus control region and the human growth hormone poly(A). HDAd was produced in 116 cells with the helper virus AdNG163 as described in detail elsewhere41,42. The generation of the HDAd vector expressing CRE recombinase under the control of the APOA1 promoter was previously described43. Hepatic transduction was achieved by intravenous administration (retro-orbital) of approximately 400 ml corresponding to 2×1012 viral particles per mouse. In co-infection experiments, 1×1012 viral particles per mouse of each virus was used. Only inbred C57BL/6 male mice were used in the HDAd-TFEB infection experiments. Age and sex-matched mice infected with a transgeneless HDAd vector were controls.
Histology and electron microscopy.
Livers were dissected, post-fixed with buffered 4% paraformaldehyde overnight at 4°C, cryoprotected and embedded in OCT. Cryostat sections of 10 μm were cut. Haematoxylin/eosin and oil red O staining protocols were performed following the IHC world protocols. Ultrathin sections were stained with Reynold’s lead citrate for 4 min, air-dried and examined on a Hitachi H7500 transmission electron microscope. Images were captured using a Gatan US1000 digital camera and Digital Micrograph v1.82.366 software.
C. elegans electron microscopy was performed as follows: wild-type and hlh-30(tm1978) synchronized L4 populations were transferred from nematode-growth media (NGM) + Escherichia coli OP50 to NGM plates supplemented with kanamycin (100 μg ml−1) and with E. coli OP50 for the control fed condition or in the absence of food for the starvation conditions. Animals were collected and incubated in fixation buffer (2.5% glutaraldehyde and 1.0% paraformaldehyde in 0.05 M sodium cacodylate buffer, at pH 7.4 plus 3.0% sucrose) 24 h after starvation at 25 °C. During the initiation of fixation, animals were cut in half with a surgical blade in a drop of fixative under a dissecting microscope, fixed overnight at 4 °C, rinsed in 0.1 M cacodylate buffer, post-fixed in 1.0% osmium tetroxide 0.1 M cacodylate buffer, rinsed in buffer and water. Animals were embedded in 2% agarose in phosphate buffered saline (PBS), dehydrated through a graded series of ethanol washes to 100%, then 100% propylene oxide, and finally 1:1 propylene oxide/EPON overnight. Blocks were infiltrated in 100% EPON and then embedded in fresh EPON overnight at 60 °C. Thin sections were cut on a Leica UC6 ultramicrotome and collected on formvar-coated grids. Sections were post-stained with uranyl acetate and lead citrate and viewed using a JEOL 1011 transmission electron microscope at 80 kV with an AMT digital imaging system (Advanced Microscopy Techniques).
Glucose, insulin and pyruvate tolerance tests.
In vivo metabolic studies were performed using the Oxymax Columbus Instruments Comprehensive Lab Animal Monitoring System. For glucose tolerance tests, mice were injected with glucose (1.5 mg g−1 body weight) after 6 h fasting. For insulin tolerance tests, 4 h fasted mice were injected (intraperitoneal) with insulin (0.75 milliunit g−1 body weight, Humulin R; Eli Lily). Experiments were performed between 10:00 and 12:00. Blood was drawn 15, 30, 60 and 120 min after treatment for determination of glucose and insulin levels. Blood glucose concentrations were measured through tail bleed before and at the times indicated after injection.
Tissue lipid quantification.
Liver triglycerides, free fatty acids and cholesterol were extracted as follows: briefly, pulverized liver was homogenized in PBS, then extracted using chloroform/methanol (2:1), dried overnight and re-suspended in a solution of 60% butanol 40% Triton X-114/methanol (2:1). Measurements were normalized to protein content in the initial homogenate by DC protein assay (Bio-Rad).
Plasma chemistry analysis.
Blood was collected from the orbital plexus under isoflurane (Vedco) anaesthesia. Plasma was frozen in aliquots at −20 °C or used immediately after collection. Specific enzymatic kits were used for determination of serum non-esterified fatty acids, ketone bodies (Wako), glycerol (Sigma Aldrich), cholesterol and triglycerides (Infinity). Plasma glucose was monitored by a glucometer. Insulin was measured by ELISA (Mercodia).
Body composition analysis.
Whole-body compositions of mice were analysed by EchoMRI (Echo Medical Systems), according to the manufacturer’s instructions.
Fatty acid oxidation measurement.
The increased oxygen consumption rate following BSA-conjugated palmitate addition was measured using the XF24 extracellular flux analyser (Seahorse Bioscience) following the manufacturer’s instructions.
In vivo metabolic analysis.
Analysis of oxygen consumption (), carbon dioxide consumption () and respiratory exchange ratio was performed using the Oxymax Columbus Instruments Comprehensive Lab Animal Monitoring System; mice were acclimatized to the system for 24 h before data collection. All of the data were normalized to weight. Rates of carbohydrate () and fat () utilization were calculated as previously described44,45.
TFEB antibodies.
To recognize both human and mouse TFEB, the mouse anti-TFEB monoclonal antibody was purchased from Myr Biosource (catalogue No. MBS120432). To recognize only the human TFEB isoform we used the rabbit anti-TFEB polyclonal antibody from Cell Signaling (# 4240).
Cell cultures.
For hepatocytes, two-month-old mice were deeply anaesthetized with Avertin (240 mg kg−1) and perfused first with 25 ml of HBSS (Sigma H6648) supplemented with 10 mM HEPES and 0.5 mM EGTA and after with a similar solution containing 100 U ml−1 of collagenase (Wako) and 0.05 mg ml−1 of trypsin inhibitor (Sigma). Liver was dissociated in a Petri dish and the cell pellet was washed in HBSS once and in a 50% Percoll gradient (Perkin Helmer) and plated at 5×105 cells per 35 mm dish. Cells were cultured in William’s medium E supplemented with 10% FBS, 2 mM glutamine, 0.1 mM insulin, 1 mM dexamethasone and penicillin/streptomycin. For starvation experiments, 24 h after preparation, cells were incubated with HBSS (+CaCl2 and MgSO4; GIBCO) supplemented with 10 mM HEPES (GIBCO) and cultured for 24 h before collection.
MEFs were isolated from embryonic day (E)13.5 embryos following previously established protocols.
RNA extraction, quantitative PCR and statistical analysis.
Total RNA was first extracted from cells using TRIzol (Invitrogen) and re-purified with a RNeasy column (Qiagen). Reverse transcription was performed using TaqMan reverse transcription reagents (Applied Biosystems). Lysosomal and autophagic gene-specific primers were previously reported29. Lipid metabolism gene-specific primers are listed in Supplementary Table S10. Fold change values were calculated using the ΔΔCt method29. An unpaired t-test was used to calculate statistical significance.
Bioinformatics and data visualization.
Venn diagrams and pie charts were generated using custom annotation scripts.
Microarray data processing.
Gene expression analysis was performed on two-month-old male mice of the indicated genotype as previously reported11,29. The data have been deposited in NCBIs Gene Expression Omnibus46 (GEO) and are accessible through GEO Series accession numberGSE35015 (overTFEB data set) andGSE36510 (WT_starved data set). Low-level analysis to convert probe level data to gene level expression was performed using the Robust Multiarray Average (RMA) function of the Bioconductor project47.
Statistical analysis of differential gene expression.
For each gene, a Bayesian t-test (Cyber-t; ref. 48) was used on RMA normalized data to determine whether there was a significant difference in expression between mice overexpressing TFEB (injected mice) versus non-injected mice used as the control (GSE35015—overTFEB data set); the same analysis was repeated to compare expression in starved wild-type mice versus wild-type mice (GSE36510 -WT_starved data set). P-value adjustment for multiple comparisons was done with the FDR of Benjamini–Hochberg49. The threshold for statistical significance chosen was FDR≤0.05; further filtering was performed by selection genes with an absolute fold change ≥1.5 for both increased (upregulated genes) and decreased (downregulated genes) expression levels. In the overTFEB data set, we selected 1,814 probesets corresponding to 1,384 differentially expressed genes (GSE35015). In the WT_starved data set we selected 467 probesets corresponding to 370 differentially expressed genes (GSE36510).
Microarray data analysis.
For each microarray data set, the Gene Ontology Enrichment Analysis13,14 (GOEA) was performed on the upregulated and downregulated gene lists, separately, by using the DAVID online tool (DAVID Bioinformatics Resources 6.7) restricting the output to all Biological Process terms (BP_ALL) (refer to Supplementary Tables S2 and S3 for the overTFEB data set; refer to Supplementary Table S5 for the 132 genes differentially expressed both in WT_starved and in overTFEB data sets listed in Supplementary Table S4). The threshold for statistical significance of GOEA was FDR≤10% and enrichment score ≥1.5. In Fig. 1 the list of 1,814 probesets (GSE35015) was mapped on the GO hierarchy50,51 rooted in the Lipid Metabolic Process (GO:0006629) term. We considered the sub-categories of the two main sub-categories of the Lipid Metabolic Process: the lipid catabolic process (GO:0016042) and the lipid biosynthetic process (GO:0008610). The lipid catabolic process (GO:0016042) contains 6 sub-categories, and the lipid biosynthetic process (GO:0008610) contains 15 sub-categories. The cellular lipid catabolic process (GO:0044242), which is one of the sub-categories of the lipid catabolic process, was divided into 7 further sub-categories for the analysis. The two sub-categories of the lipid oxidation (GO:0034440) were also analysed. We introduced one more sub-category, the fatty acid binding and transport, to map genes as indicated in the literature. The 1,384 differentially expressed genes were mapped to 20 sub-categories (http://www.geneontology.org/; refs 50, 51): 7 sub-categories of the lipid catabolic process, 11 sub-categories of the lipid biosynthetic process, and 1 sub-category each of the lipid oxidation and the fatty acid binding and transport. The total number of the mapped genes was 124 (Supplementary Table S7). The distribution of these genes among the 20 sub-categories was visualized in Cytoscape52. The threshold for statistical significance chosen for the comparison between the overTFEB data set and the WT_starved data set was FDR≤0.05 and absolute fold change ≥1.5: we thus selected 162 probesets corresponding to 132 differentially expressed genes (Supplementary Table S4). The GOEA on the list of 132 genes was performed as described above and reported in Supplementary Table S5. The total number of genes, out of 132, mapped to the different Lipid Metabolic Process sub-categories was 52 (Supplementary Table S6).
ChIP.
Mice overexpressing TFEB–FLAG in liver were previously described11. Transgeneless littermates were used as controls. Livers were isolated from 5-day-old mice and incubated in 1% formaldehyde for 10 min at room temperature to crosslink DNA to associated proteins. Tissues were washed twice with ice-cold PBS containing 0.125 M glycine and protease inhibitors (Complete protease inhibitor cocktail tablets, Roche Applied Science) and then homogenized in lysis buffer (10 mM Tris–HCl at pH 7.5, 10 mM NaCl, 3 mM MgCl2 and 0.5% NP40) containing protease inhibitors. After 10 min of incubation on ice, lysates were centrifuged for 5 min at 1,000g and 4 °C, and the nuclear pellets sonicated with a Branson Sonifier 450 for 6 s at power level 6. Homogenates were then digested with 300 U of micrococcal nuclease for 5 min at 37 °C (Worthington Biochemical Corporation) to generate fragments with an average length of ∼ 100–200 base pairs (bp), as determined empirically by agarose gel electrophoresis of the fragmented chromatin sample. For immunoprecipitation, 200 μl of chromatin was diluted 1:10 in ChIP dilution buffer (Millipore Corporation) and 1% of the diluted sample was set aside for input. The sample was precleared with streptavidin beads (Thermo Scientific) at 4 °C for 3 h. The precleared lysate was incubated overnight with 1 μg of biotinylated anti-FLAG antibody (BioM2 SIGMA). After immunoprecipitation, the beads were washed at room temperature (for 5 min each) with 700 μl of low-salt buffer, high-salt buffer, LiCl buffer (Millipore Corporation) and TE buffer (10 mM Tris–HCl at pH 7.4 and 1 mM EDTA at pH 8.0). Elution was performed twice in 250 μl of fresh elution buffer (1% SDS and 0.1 M NaHCO3) for 15 min at room temperature. Eluates were combined, 20 μl of 5 M NaCl was added, and samples were incubated overnight at 65 °C to reverse the crosslinks. Input DNA was diluted in freshly made elution buffer to a volume of 500 μl, and crosslink reversal was performed. After crosslink reversal, samples were digested with 20 μg of proteinase K for 1 h at 55 °C, and DNA was recovered by standard methods in 30 μl of 10 mM Tris–HCl at pH 8.0. One μl of DNA was used for each quantitative rtPCR reaction. The quantitative rtPCR data were analysed as follows: relative proportions of immunoprecipitated DNA were determined based on the threshold cycle (Ct) value for each PCR reaction. To control for variation between ChIP fractions, for every gene promoter studied, a ΔCt value was calculated for each sample (WT, TFEB–FLAG) by subtracting the Ct value for the input (CtInput) from the Ct value for the immunoprecipitated sample (Ctantibody). As the input DNA fraction represents only 1% of the total material (see above), the CtInput value was first adjusted for this dilution factor by subtracting 6.644 cycles (log2 of 100). The formula used was: ΔCt = [Ct elute−(CtInput−6.644)]. Next, a ΔΔCt value was calculated for each sample by subtracting the ΔCtWT (transgeneless) from the corresponding ΔCt (transgene). Fold differences between the specific immunoprecipitation and the background were then determined by raising 2 to the −ΔΔCt power.
Phylogenetic analysis of HLH-30.
For identification of TFEB homologues in C. elegans, the TFEB RefSeq file (accession NP_001161299.1) was BLASTed using the NCBI BLASTP tool (http://blast.ncbi.nlm.nih.gov). BLAST results were visualized with the Distance Tree tool on the same website, using the Neighbor joining method at a cutoff sequence difference of 0.85. The resulting tree was rendered and edited in FigTree (http://tree.bio.ed.ac.uk/software/figtree).
C. elegans strains and growth.
The wild-type Bristol (N2) was obtained from the Caenorhabditis Genetics Center (CGC, University of Minnesota). hlh-30(tm1978) mutant, a gift from V.R. Ambros (University of Massachusetts Medical School, USA) was backcrossed 3 times into the wild-type background. C. elegans was grown on NGM plates seeded with E. coli OP50 at 15–20 °C according to standard procedures53.
Quantitative rtPCR analysis and statistics in C. elegans.
Synchronized fed L4 C. elegans animals were transferred from NGM with E. coli OP50 to NGM plates supplemented with kanamycin (100 μg ml−1) in the absence of food for starvation or seeded with E. coli OP50 for fed control. For re-feeding after starvation, 500 μl of 10× concentrated E. coli OP50 overnight culture was added to the starvation plate versus 500 μl of vehicle (Luria-Bertani (LB) Broth) for starvation control. cDNA was subjected to quantitative rtPCR analysis as described previously54 and data analysis was performed using the Pfaffl method55. Expression values were normalized against the control gene snb-1 and pan-act house-keeping genes, which did not vary under the conditions being tested. Statistics were performed using a two-sample two-sided unpaired t-test on ΔCt values from 3 independent biological replicates. A P value equal to or less than 0.05 was considered statistically significant.
Oil red O staining and quantification in C. elegans.
Wild-type and hlh-30 adult animals were washed twice in M9 buffer and plated onto NGM plates supplemented with kanamycin (100 μ ml), and with OP50 or without any food source for the starvation condition. After 8 h incubation at 25 °C, animals were washed twice in PBS, 0.01% Triton X-100, resuspended in dehydrating buffer (PBS, 60% isopropanol, 0.01% Triton X-100). After 15 min of incubation at room temperature with gentle shaking, animals were resuspended into oil red O dye solution (as described in ref. 56) and incubated overnight with gentle shaking. The next day, dye was removed and animals were washed twice in 1 ml of PBS, 0.01% Triton X-100, mounted between a glass slide and a coverslip, and imaged using the ×10 objective in a Nikon Eclipse E800 microscope. Oil red O staining was quantified using ImageJ from at least 30 animals per condition. Experiments have been performed three times.
Lifespan and survival statistics.
For lifespan assays, a total of 25–35 synchronized L4 hermaphrodites were transferred to each of three replicate NGM plates per strain and grown at 25 °C. NGM plates were supplemented with kanamycin (100 μg ml−1) and seeded with E. coli OP50 for the control fed condition, in the absence of any food source for the starvation condition. Once a day, animals were scored as alive or dead by gentle prodding with a platinum wire. Animals that died from crawling off the agar were excluded from the study. Kaplan–Meier statistical analyses were performed using software Prism 5 (GraphPad, http://www.graphpad.com). Survival data were compared using the log-rank test; error bars correspond to standard error (Greenwood’s method). Experiments were performed twice.
L1 arrest assay.
Bleached eggs were resuspended in M9 buffer and incubated for 24 h at room temperature. To control for the non-starved condition, a volume containing approximately ∼ 100 eggs was plated in triplicate directly after preparation onto NGM + OP50 plates and eggs were grown at 15 °C. After 24 h of starvation, the same volume was plated in triplicate on NGM + OP50 and incubated at 15 °C. After 2 to 3 days, L4 and adult animals from non-starved and starved conditions were counted. Data are representative of 3 independent replicates.
Change history
25 June 2013
In the version of this Article originally published, in the Methods section, Supplementary Table S8 was incorrectly cited as listing lipid metabolism gene-specific primers under the heading 'RNA extraction, quantitative PCR and statistical analysis'. The list of gene-specific primers has now been uploaded as Supplementary Table S10 and the citation in the Methods has been amended. This has been corrected in the PDF and HTML versions of this Article.
References
Bauer, M. et al. Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol. Genomics 17, 230–244 (2004).
Sokolovic, M. et al. The transcriptomic signature of fasting murine liver. BMC Genomics 9, 528 (2008).
Hakvoort, T. B. et al. Interorgan coordination of the murine adaptive response to fasting. J. Biol. Chem. 286, 16332–16343 (2011).
Finn, P. F. & Dice, J. F. Proteolytic and lipolytic responses to starvation. Nutrition 22, 830–844 (2006).
Fontana, L., Partridge, L. & Longo, V. D. Extending healthy life span–from yeast to humans. Science 328, 321–326 (2010).
Zinke, I., Schutz, C. S., Katzenberger, J. D., Bauer, M. & Pankratz, M. J. Nutrient control of gene expression in Drosophila: microarray analysis of starvation and sugar-dependent response. EMBO J. 21, 6162–6173 (2002).
Van Gilst, M. R., Hadjivassiliou, H. & Yamamoto, K. R. A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc. Natl Acad. Sci. USA 102, 13496–13501 (2005).
Mizushima, N. Autophagy: process and function. Genes Dev. 21, 2861–2873 (2007).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009).
Singh, R. & Cuervo, A. M. Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504 (2011).
Yang, L., Li, P., Fu, S., Calay, E. S. & Hotamisligil, G. S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478 (2010).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011).
Palmieri, M. et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 (2011).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Finck, B. N. & Kelly, D. P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116, 615–622 (2006).
Spiegelman, B. M. & Heinrich, R. Biological control through regulated transcriptional coactivators. Cell 119, 157–167 (2004).
Vega, R. B., Huss, J. M. & Kelly, D. P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 20, 1868–1876 (2000).
Lin, J., Handschin, C. & Spiegelman, B. M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 (2005).
Komatsu, M. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 (2005).
Komatsu, M. et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 (2007).
Bordone, L. et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 6, 759–767 (2007).
Vaisse, C. et al. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 14, 95–97 (1996).
Lai, C. H., Chou, C. Y., Ch’ang, L. Y., Liu, C. S. & Lin, W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 10, 703–713 (2000).
Grove, C. A. et al. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138, 314–327 (2009).
Kaeberlein, T. L. et al. Lifespan extension in Caenorhabditis elegans by complete removal of food. Aging Cell 5, 487–494 (2006).
Johnson, T. E., Mitchell, D. H., Kline, S., Kemal, R. & Foy, J. Arresting development arrests ageing in the nematode Caenorhabditis elegans. Mech. Ageing Dev. 28, 23–40 (1984).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 1095–1108 (2012).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Roczniak-Ferguson, A. et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42 (2012).
Becskei, A., Seraphin, B. & Serrano, L. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 20, 2528–2535 (2001).
Siciliano, V. et al. Construction and modelling of an inducible positive feedback loop stably integrated in a mammalian cell-line. PLoS Comput. Biol. 7, e1002074 (2011).
Kielbasa, S. M. & Vingron, M. Transcriptional autoregulatory loops are highly conserved in vertebrate evolution. PLoS One 3, e3210 (2008).
Handschin, C., Rhee, J., Lin, J., Tarr, P. T. & Spiegelman, B. M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1α expression in muscle. Proc. Natl Acad. Sci. USA 100, 7111–7116 (2003).
Tsunemi, T. et al. PGC- 1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 4, 142ra197 (2012).
Bostrom, P. et al. A PGC1- α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468 (2012).
Badman, M. K. et al. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 (2007).
Inagaki, T. et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 (2007).
Beale, E. G., Clouthier, D. E. & Hammer, R. E. Cell-specific expression of cytosolic phosphoenolpyruvate carboxykinase in transgenic mice. FASEB J. 6, 3330–3337 (1992).
Palmer, D. & Ng, P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 8, 846–852 (2003).
Ng, P., Parks, R. J. & Graham, F. L. Preparation of helper-dependent adenoviral vectors. Methods Mol. Med. 69, 371–388 (2002).
Li, R. et al. Gene therapy targeting LDL cholesterol but not HDL cholesterol induces regression of advanced atherosclerosis in a mouse model of familial hypercholesterolemia. J Genet Syndr. Gene Ther. 2, 106 (2011).
Hawley, S. A. et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 8, 918–922 (2012).
Couture, S., Massicotte, D., Lavoie, C., Hillaire-Marcel, C. & Peronnet, F. Oral [(13)C]glucose and endogenous energy substrate oxidation during prolonged treadmill running. J. Appl. Physiol. 92, 1255–1260 (2002).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Baldi, P. & Long, A. D. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17, 509–519 (2001).
Klipper-Aurbach, Y. et al. Mathematical formulae for the prediction of the residual β cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med. Hypotheses 45, 486–490 (1995).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Carbon, S. et al. AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Irazoqui, J. E., Ng, A., Xavier, R. J. & Ausubel, F. M. Role for β-catenin andHOX transcription factors in Caenorhabditis elegans and mammalian hostepithelial-pathogen interactions. Proc. Natl Acad. Sci. USA 105, 17469–17474 (2008).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).
O’Rourke, E. J., Soukas, A. A., Carr, C. E. & Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10, 430–435 (2009).
Acknowledgements
We thank G. Karsenty, D. Moore, H. Zoghbi, S. Colamarino, E. Abrams, D. Sabatini and R. Zoncu for critical reading of the manuscript. We thank G. Diez-Roux for critical reading of the manuscript, helpful discussions and support in manuscript preparation. We are also grateful to D. Medina, C. Spampanato and F. Annunziata for their contribution. We thank A. Soukas for help with the oil red O staining of C. elegans. We acknowledge the support of the Italian Telethon Foundation grant numbers TGM11CB6 (C.S., R.D.C. and A.B) and TGM11SB1 (A.C. and D.D.B.); the Beyond Batten Disease Foundation (C.S., F.V., T.H. and A.B.); European Research Council Advanced Investigator grant no. 250154 (A.B.); March of Dimes #6-FY11-306 (A.B.); US National Institutes of Health (R01-NS078072) (A.B.). This work was supported in part by grants from the US National Institutes of Health (R01-HL51586) to L.C. and the Diabetes and Endocrinology Research Center (P30-DK079638, L.C.) and the Mouse Metabolism Core (P.K.S.) at Baylor College of Medicine. T.J.K. is in part supported by the Cancer Prevention and Research Institute of Texas (RP110390). O.V. was funded by a Fund for Medical Discovery postdoctoral fellowship from the Massachusetts General Hospital. This study was funded in part by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM101056-01 to J.E.I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
C.S., G.M., P.K.S., F.V., O.V., T.H., R.D.C., A.C., D.P., T.J.K. and A.C.W. performed the experiments. D.D.B. supervised bioinformatic analyses. J.E.I. supervised the C. elegans experiments, L.C. supervised the metabolic studies, and A.B. and C.S. designed the overall study and supervised the work. All authors discussed the results and made substantial contributions to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 1219 kb)
Supplementary Information
Supplementary Table 1 (XLSX 59 kb)
Supplementary Information
Supplementary Table 2 (XLSX 110 kb)
Supplementary Information
Supplementary Table 3 (XLSX 87 kb)
Supplementary Information
Supplementary Table 4 (XLS 63 kb)
Supplementary Information
Supplementary Table 5 (XLS 57 kb)
Supplementary Information
Supplementary Table 6 (XLSX 39 kb)
Supplementary Information
Supplementary Table 7 (XLSX 50 kb)
Supplementary Information
Supplementary Table 8 (XLS 162 kb)
Supplementary Information
Supplementary Table 9 (XLSX 51 kb)
Supplementary Information
Supplementary Table 10 (XLS 10 kb)
Rights and permissions
About this article
Cite this article
Settembre, C., De Cegli, R., Mansueto, G. et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15, 647–658 (2013). https://doi.org/10.1038/ncb2718
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb2718
