
Abstract
In contrast to infections with human immunodeficiency virus (HIV) in humans and simian immunodeficiency virus (SIV) in macaques, SIV infection of a natural host, sooty mangabeys (Cercocebus atys), is non-pathogenic despite high viraemia1. Here we sequenced and assembled the genome of a captive sooty mangabey. We conducted genome-wide comparative analyses of transcript assemblies from C. atys and AIDS-susceptible species, such as humans and macaques, to identify candidates for host genetic factors that influence susceptibility. We identified several immune-related genes in the genome of C. atys that show substantial sequence divergence from macaques or humans. One of these sequence divergences, a C-terminal frameshift in the toll-like receptor-4 (TLR4) gene of C. atys, is associated with a blunted in vitro response to TLR-4 ligands. In addition, we found a major structural change in exons 3–4 of the immune-regulatory protein intercellular adhesion molecule 2 (ICAM-2); expression of this variant leads to reduced cell surface expression of ICAM-2. These data provide a resource for comparative genomic studies of HIV and/or SIV pathogenesis and may help to elucidate the mechanisms by which SIV-infected sooty mangabeys avoid AIDS.
Similar content being viewed by others

Main
SIV infection of natural hosts, such as sooty mangabeys, is typically non-pathogenic despite high viraemia. This is in stark contrast to HIV infection in humans and experimental SIV infection in rhesus macaques (Macaca mulatta) that progress to AIDS unless treated with antiretroviral therapy. The main virological and immunological features of natural SIV infection in sooty mangabeys have been described over the past 15 years in studies that compared and contrasted this infection with the pathogenic infections of HIV and SIV in humans and rhesus macaques1. SIV-infected sooty mangabeys show several features that have been observed in pathogenic infections, including high viraemia, short in vivo lifespan of productively infected cells, depletion of mucosal CD4+ T cells, strong type-I interferon response in the acute infection, and cellular immune responses that fail to control virus replication. However, in contrast to pathogenic infections, SIV-infected sooty mangabeys (i) have healthy CD4+ T cell levels; (ii) do not experience mucosal immune dysfunction, avoiding depletion of T helper 17 (TH17) cells, intestinal epithelial damage and microbial translocation; (iii) maintain low levels of immune activation during the chronic infection; and (iv) achieve compartmentalization of virus replication that preserves central-memory and stem-cell memory CD4+ T cells as well as follicular TH cells1,2. An additional notable feature of SIV infection in natural hosts is the low rate of mother-to-infant transmission that is related to low expression of CCR5 on circulating and mucosal CD4+ T cells3. Although many aspects of the natural course of SIV infection in sooty mangabeys have now been described, the key molecular mechanisms by which these animals avoid AIDS remain poorly understood.
In this study, we sequenced the genome of a captive sooty mangabey and compared this genome to the genomes of AIDS-susceptible primates to look for candidate genes that may influence susceptibility to AIDS in SIV-infected hosts. We sequenced genomic DNA to a whole-genome coverage of about 180× using the Illumina HiSeq 2000 platform, and produced an initial assembly using ALLPATHS-LG, Atlas-Link and Atlas-GapFill (see Methods for details). The total size of the assembled C. atys genome (Caty_1.0; NCBI accession number GCA_000955945.1) is around 2.85 Gb, with a contig N50 size of 112.9 kb and scaffold N50 size of 12.85 Mb (Table 1). Genome annotation identified 20,829 protein-coding genes and 4,464 non-coding genes in the C. atys assembly, which is comparable to other available draft quality genomes of nonhuman primates (Table 1). These analyses demonstrate that the Caty_1.0 reference genome is of sufficient quality to facilitate population-scale whole-genome and transcriptome sequencing studies.
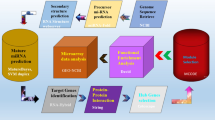
To identify novel immunogenetic factors specific to C. atys that may be involved in the ability of this species to avoid progression to AIDS, we established a bioinformatic pipeline for a comparative protein analysis (Fig. 1 and Extended Data Fig. 1, see Methods for details). Using this approach, we found 34 candidate immune-related genes with sequences that diverged between C. atys and M. mulatta (Table 1 and Extended Data Table 1). Although we cannot exclude a role of immune genes with minor differences in C. atys and M. mulatta, the highly divergent genes listed in Table 1 and Extended Data Table 1 constitute candidate genes involved in the outcomes of SIV infection in these two species.

(1) Sooty mangabey (SM) orthologues were selected by BLAST alignment of C. atys NCBI protein predictions (blue) to curated rhesus macaque (RM) protein models (green22) and alignment scores were calculated. (2) NCBI transcript predictions with RNA-seq support were identified by BLAT alignment of de novo assembled C. atys RNA-seq transcripts (orange) to C. atys NCBI coding sequence (CDS) predictions (red). (3) Subsquently, corresponding RNA-seq-supported C. atys NCBI protein predictions were selected. (4) C. atys proteins with high similarity (>97% identity) to M. mulatta proteins were filtered out. (5) Immune genes according to Gene Ontology (GO) term classification (immune response) were chosen for further analysis and (6) confirmed by manual inspection.
Our screen identified sequence divergence in a number of proteins that are important during HIV infection, such as APOBEC3C (91.6%) and BST2 (also known as tetherin, 95.1%), as well as pattern-recognition receptors (MBL2, CLEC4A, CLEC4D and CLEC6A), the antiviral sensor cyclic GMP–AMP synthase (cGAS (also known as MB21D1)) and other immune mediators (Extended Data Table 1). Because CD4 and CCR5 are important for AIDS pathogenesis, we aligned the sequences of CaCD4 and CaCCR5 to MmCD4 and MmCCR5, respectively4,5. Neither gene showed any major structural changes in the wild-type variants, although CD4 was slightly below the 97% threshold of identity (Extended Data Fig. 1b, c). In addition, we found specific gene families in C. atys that are expanded relative to M. mulatta, humans and other primates (Extended Data Table 2a). Notably, we detected localized regions of increased substitution, defined by a clustered difference of three or more amino acids, in 10 genes. The most marked variations in the amino acid sequence of C. atys compared to M. mulatta were observed in ICAM-2 and TLR-4 (Table 1).
ICAM-2 is an approximately 60-kDa transmembrane glycoprotein of the immunoglobulin superfamily, which is expressed on various immune cells and implicated in lymphocyte homing and recirculation6. ICAM-2 ligands are lymphocyte function-associated antigen-1 and the C-type lectin DC-SIGN7. We discovered a misalignment of the ICAM-2 proteins between C. atys and M. mulatta that starts in exon 3 (Extended Data Fig. 2a). This difference is explained by a 499-bp deletion starting from exon 3 of CaICAM2, as detected by PCR and Sanger sequencing (Fig. 2a and Extended Data Fig. 3). We subsequently confirmed the expression of this truncated form of ICAM-2 in ten out of ten additional C. atys genome sequences (Extended Data Fig. 2b). By contrast, analysis of the whole-genome sequences of 15 baboons and more than 130 rhesus macaques demonstrated that only the full-length ICAM-2 protein was found in all individuals (data not shown)8. The ICAM-2 deletion may be specific to C. atys, as it is not present in any other known primate sequences, including other natural SIV hosts, such as the African green monkey, drill and colobus monkey. Transcript models generated from de novo assembled C. atys RNA-sequencing (RNA-seq) data from 14 different tissues showed that the mature mRNA sequence of CaICAM2 retains substantial portions of what is part of the intronic sequence in other nonhuman primates, and thus codes for a markedly different final gene product (Extended Data Figs 2, 3). Splice-junction sequence analysis showed intact splicing for all four exons in M. mulatta, but no splice junctions were found between exons 3 and 4 in C. atys, indicating severe splicing defects due to the deletion (Extended Data Fig. 4).

a, PCR to confirm a putative 0.5-kb deletion in the CaICAM2. b, ICAM-2 surface expression of primary CD4+ cells by flow cytometry. n = 3; representative plots for c. c, ICAM-2 surface expression in B cells, CD4+ and CD8+ T cells from human, rhesus macaques and sooty mangabeys. n = 3 biologically independent samples for each species. d, ICAM-2-specific western blot using peripheral blood mononuclear cells from M. mulatta and C. atys. n = 3 M. mulatta; n = 2 C. atys; one representative biological sample per species is shown. For gel source data, see Supplementary Figs 1, 3.
To test whether the observed genetic difference in ICAM2 has functional consequences, we measured ICAM-2 surface expression on immune cells from humans, M. mulatta and C. atys with an antibody that recognizes a conserved epitope between these species9. ICAM-2 was readily detected on T cells and B cells from humans and M. mulatta, but not from C. atys (Fig. 2b, c), suggesting that ICAM-2 is not functional in lymphocytes of C. atys. However, a truncated, lower molecular weight form of ICAM-2 could be detected intracellularly by western blot in C. atys cells (Fig. 2d), thus demonstrating the presence of the predicted truncated ICAM-2 protein. Overall, these data indicate that the presence of a species-specific gene sequence difference in CaICAM2 results in the abrogation of surface expression of this protein in C. atys. Further studies are needed to elucidate potential links between this truncated form of ICAM-2 and the remarkable immunological features of SIV infection in this species.
TLR-4 is a pattern recognition receptor that senses lipopolysaccharides (LPS) on gram-negative bacteria and initiates pro-inflammatory cytokine induction, maturation and activation in macrophages, dendritic cells and other immune cells. During pathogenic HIV or SIV infections, exacerbated TLR-4 stimulation and concomitant pro-inflammatory signalling elicited by microbial translocation is considered a primary mechanism that underlies HIV-induced chronic immune activation10,11. Here, we found that the TLR-4 protein sequences of M. mulatta and C. atys are markedly different at the C terminus (Extended Data Fig. 5a). We confirmed the underlying difference in the TLR4 nucleotide sequence by Sanger sequencing (Extended Data Fig. 5b, c). We next analysed the genomic DNA sequence of TLR4 in 10 additional sooty mangabeys and found that the observed DNA sequence difference was present in all individuals (Extended Data Fig. 6a). Alignment of TLR-4 protein sequences from different primate species revealed that the 17-amino-acid longer C-terminal sequence is only found in natural SIV hosts, such as African green monkey, drill and colobus monkey (Fig. 3a), whereas non-natural hosts, including M. mulatta and baboons show expression of the short TLR-4 C-terminal sequence.

a, Alignment of C-terminal TLR-4 protein sequences from different primate species (starting at human TLR-4 amino acid position 821). b, Primate phylogenetic tree with colour-coding according to the TLR-4 C terminus as indicated in a. Phylogeny appears as in ref. 14. c, Cytokine release from blood of rhesus macaques (n = 9 biologically independent samples) and sooty mangabeys (n = 8 biologically independent samples) after LPS stimulation as measured by cytometric bead array. d, mRNA expression in whole blood after LPS stimulation quantified by quantitative PCR (qPCR). n = 4 biologically independent samples for each species. e, TNF and IL-6 cytokine release from blood of rhesus macaques and sooty mangabeys over the course of SIV infection. n = 5 biologically independent samples for each species. Data are mean ± s.d. (c–e), unpaired two-sided Student’s t-test, P values are indicated (c, d). f, Gene set enrichment analysis of LPS-stimulated monocytes of rhesus macaques and sooty mangabeys using the TNF signalling via NF-κB hallmark gene set. g, GSEA of LPS-stimulated monocytes of rhesus macaques and sooty mangabey using the IL6 JAK–STAT3 hallmark gene set. h, NF-κB response to LPS of primate TLR4 variants in transfected HEK293T cells. NF-κB firefly-luciferase signals were normalized to Gaussia luciferase signals, and the relative increase in NF-κB activity compared to unstimulated controls (100%) was calculated. Data are mean ± s.e.m. of n = 5 independent experiments performed in triplicate transfections are shown. Unpaired two-sided Student’s t-test, P values are indicated. For source data of the animal studies, see Supplementary Table 1. RM SM-CT, MmTLR-4 with the C terminus of CaTLR-4; SM RM-CT, CaTLR-4 with the C terminus of MmTLR-4.
The divergence of TLR-4 amino acid sequences amongst Old World primates shows an interesting pattern of molecular evolution. First, the genomic sequence encoding the TLR4 C terminus is defined by a 1-bp deletion causing a frame shift in all Old World monkeys, both natural and non-natural hosts, including colobine and cercopithecine lineages, but it is not found in either hominoids (apes and humans) or platyrrhines (New World monkeys) (Extended Data Fig. 6b). This suggests that this mutation occurred after the hominoid–Old World monkey divergence approximately 25 million years ago12. Second, there is a G-to-A nucleotide substitution in the non-natural host Old World monkeys (baboons and macaques) that creates a truncated protein in these species8 (Extended Data Fig. 6b). Although a naive analysis of this pattern would suggest two independent mutational changes in TLR4, the short internal branch of the species tree implies that incomplete lineage sorting of an ancestral polymorphism could also generate this pattern13 (Fig. 3b). To test this hypothesis, we examined the TLR4 gene tree among 17 primate species. While generally supporting the relationships among these species (Fig. 3b), the analysis also found a number of nucleotide positions—spaced throughout the gene—consistent with incomplete lineage sorting between C. atys, baboon and M. mulatta (Extended Data Fig. 7). The incomplete lineage sorting hypothesis is also more likely, given that balancing selection is often found to be acting on immune-related genes. Therefore, even though baboons are believed to be more closely related to sooty mangabeys and drills than to rhesus macaques, the phylogeny of Old World monkeys is compatible with the possibility of a single G-to-A mutation creating the truncated form of the protein in the common ancestor of baboons, rhesus macaques and sooty mangabeys12,14 (Fig. 3b).
We next investigated potential differences in TLR-4 function between M. mulatta and C. atys. Our previous work has shown that macrophages from C. atys exhibit higher expression of tetherin, APOBEC and TRIM5α in response to LPS compared to M. mulatta15. This is consistent with the relative resistance of C. atys macrophages to in vivo SIV infection after experimental CD4+ T cell depletion compared to SIV-infected M. mulatta macrophages16. Here we analysed cytokine gene expression and protein production after LPS stimulation, and found reduced mRNA expression and secretion of TNF (also known as TNF-α) and IL-6 in cells from C. atys compared to M. mulatta (Fig. 3c, d). Because some commercial LPS preparations contain lipoprotein contaminants that can induce TLR-2 signalling, we confirmed the TLR-4 specificity of the reduced LPS response using the selective TLR-4 agonist17 lipid-A (Extended Data Fig. 8a, b). Next, we found that the species-specific differences between C. atys and M. mulatta in LPS-induced TNF and IL-6 production were maintained in acute and chronic infection (Fig. 3e and Extended Data Fig. 8c). Additionally, we did not observe any difference in the mRNA levels of TLR4 in cells from C. atys and M. mulatta, nor did the expression of any factors in the TLR-4–MyD88–TRIF signalling axis correlate with TNF and IL-6 production (Extended Data Fig. 8d and Extended Data Table 3). To more broadly characterize the effect of attenuated TLR-4 signalling in C. atys, we performed comparative RNA-seq profiling of LPS-treated monocytes, and found lower production of CaTNF and CaIL6 mRNA (Extended Data Fig. 8e). Moreover, using gene set enrichment analysis (GSEA), we observed that induction of pro-inflammatory genes was broadly and significantly reduced in cells from C. atys (Fig. 3f, g and Extended Data Fig. 9). Overall, these results indicate that LPS stimulation of blood cells from C. atys results in a blunted production of pro-inflammatory cytokines. To establish a link between the C-terminal TLR4 sequence difference and the responsiveness to LPS, we analysed the TLR-4 orthologues of humans, C. atys and M. mulatta in an NF-κB reporter assay. We observed a significantly attenuated NF-κB response to LPS of C. atys TLR-4 (CaTLR-4) compared to M. mulatta TLR-4 (MmTLR-4). Using chimaeric constructs encoding MmTLR4 with the C terminus of CaTLR4 or CaTLR4 with the C terminus of MmTLR4, we confirmed that the TLR4 C terminus is responsible for this phenotypic difference (Fig. 3h). This demonstrates a sequence–function relationship of the TLR4 C terminus and suggests a novel mechanism contributing to the lower immune activation of SIV-infected sooty mangabeys.
Over the past decade the genomes of more than 25 nonhuman primate species have been sequenced, assembled and annotated18. This knowledge has improved our understanding of primate evolution, biology and general physiology, which has informed human biology and medicine. Here, we report a high-coverage, high-contiguity whole-genome sequence for C. atys, a natural SIV host. Comparative genomic analyses of natural and non-natural SIV hosts provide candidate genes that potentially influence susceptibility to AIDS in SIV-infected hosts. We have previously used trancriptomics to characterize the host response to SIV infection of C. atys and African green monkeys19,20. Here, we examined the mechanisms of AIDS resistance of a natural SIV host genome-wide using genome sequencing. We identified candidate genes that show sequence changes that are specific to C. atys and two gene products (ICAM-2 and TLR-4), which show structural differences between C. atys and M. mulatta that may influence cell-surface expression (ICAM-2) and downstream signalling (TLR-4) of these proteins. Our findings may also explain prior results showing that not all natural SIV hosts respond to infection in the same way, suggesting that in each primate species, multiple distinct mechanisms may contribute to the phenotype, rather than mutations in single genes, as has been purported, and eventually refuted, in other studies21. Further comparative studies with additional natural SIV host species may identify additional similarities (or differences) in the genes involved in the evolutionary pathways that led to AIDS resistance in different species of African nonhuman primates.
In this study, we used whole-genome sequencing and comparative genomic analysis to identify candidate genes regulating host resistance to AIDS. Future studies in which these candidate genes are manipulated in vivo during SIV infection are needed to characterize to what extent these genes may influence the non-pathogenic nature of SIV infection in sooty mangabeys.
Methods
Sequencing and assembly of the sooty mangabey genome
DNA from a female sooty mangabey (C. atys) born and maintained at the Yerkes National Primate Research Center was extracted from whole blood. The animal selected for sequencing was one of the original dams of a large matrilineal line of the colony. In addition, she possessed the most common MHC haplotype observed within the group. As such, her genetic constitution within the closed population was thought to be the most representative of any single animal. All animals were housed at the Yerkes National Primate Research Center of Emory University and maintained in accordance with US NIH guidelines. All studies were approved by the Emory University Institutional Animal Care and Usage Committee. Following quality control to ensure purity and molecular weight, a series of Illumina sequencing libraries were prepared using standard procedures. Paired-end libraries with nominal insert sizes 180 bp and 500 bp were produced. In brief, 1 μg of DNA was sheared to the desired size using a Covaris S-2 system. Sheared fragments were purified with Agencourt AMPure XP beads, end-repaired, dA-tailed and ligated to Illumina universal adaptors. After adaptor ligation, DNA fragments were further size selected by agarose gel and PCR amplified for six to eight cycles using Illumina P1 and Index primer pair and Phusion High-Fidelity PCR Master Mix (New England Biolabs). The final library was purified using Agencourt AMPure XP beads and quality assessed by Agilent Bioanalyzer 2100 (DNA 7500 kit) to determine library quantity and fragment size distribution before sequencing.
Long mate-pair libraries with 2-kb, 3-kb, 5-kb and 8-kb insert sizes were constructed according to the manufacturer’s protocol (Mate Pair Library v.2 Sample Preparation Guide 15001464 Rev. A Pilot Release). In brief, 5 μg (for 2- and 3-kb size libraries) or 10 μg (5- and 8-kb libraries) of genomic DNA was sheared to the desired size by Hydroshear (Digilab), then end-repaired and biotinylated. Fragment sizes between 1.8–2.5 kb (2 kb), 3.0–3.7 kb (3 kb), 4.5–6.0 kb (5 kb) or 8–10 kb (8 kb) were purified from a 1% low-melting agarose gel and circularized by blunt-end ligation. These size-selected circular DNA fragments were then sheared to 400 bp (Covaris S-2), purified using Dynabeads M-280 Streptavidin Magnetic Beads, end-repaired, dA-tailed and ligated to Illumina PE sequencing adapters. DNA fragments with adaptor molecules on both ends were amplified for 12 to 15 cycles with Illumina P1 and Index primers. Amplified DNA fragments were purified with Agencourt AMPure XP beads. Quantification and size distribution of the final library was determined as described above before sequencing.
Sequencing was performed on Illumina HiSeq 2000 instruments, generating 100-bp paired-end reads. Raw sequences have been deposited in NCBI under Bioproject PRJNA157077. Reads were assembled using ALLPATHS-LG and further scaffolded and gap-filled using in-house tools Atlas-Link (v.1.0) and Atlas GapFill (v.2.2) (https://www.hgsc.bcm.edu/software/)23. Atlas-link is a scaffolding or super-scaffolding method that uses all unused mate pairs to increase scaffold sizes and create new scaffolds in draft-quality assemblies. Those modified scaffolds are then ordered and oriented. Atlas GapFill is run on a super-scaffolded assembly. Regions with gaps are identified and reads mapping within or across those gaps are locally assembled using different assemblers (Phrap, Newbler and Velvet) in order to bridge the gaps with the most conservative assembly of previously unincorporated reads.
PBJelly (v.14.9.9) is a pipeline that improves the contiguity of draft assemblies by filling gaps, increasing contig sizes and super scaffolding by making use of long reads24. We used 12.3× coverage of long Pacific Biosciences RSI and RS II sequences, along with the gap-filled Illumina read assembly, as input into PBJelly to produce the final C. atys hybrid Illumina–PacBio assembly. This assembly is available at NCBI as Caty1.0 (RefSeq accession GCF_000955945.1).
The total size of the assembled C. atys genome is around 2.85 Gb, with a contig N50 size of 112.9 kb and scaffold N50 size of 12.85 Mb (Table 1). By comparison, this contig N50 size is greater than equivalent values for 22 of the 26 other nonhuman primate genome assemblies currently available. To assess completeness, we mapped 21,772 human protein-coding canonical transcripts to Caty_1.0 and found that 94.9% map to this C. atys genome with lengths of 95–100% (97.3% of transcripts map at length 70% or greater). As a more stringent test, we mapped 3023 Benchmarking Universal Single-Copy Orthologues (BUSCO) genes and found that over 95% are present in Caty_1.0 (88.8% complete single copy and the others present but duplicated or fragmented)25.
Genome annotation was performed through the NCBI Genome Annotation Pipeline, which generated models for genes, transcripts and proteins26. To aid accurate transcript annotation, the NCBI pipeline incorporated RNA-seq data from a sooty mangabey pooled tissue reference sample, and data from 14 separate tissues produced through a joint effort by the Nonhuman Primate Reference Transcriptome Resource (NHPRTR; http://www.nhprtr.org/)27 and the Human Genome Sequencing Center (HGSC) of Baylor College of Medicine. The NCBI process also used human RefSeq and GenBank transcripts along with other primate protein data.
Sequencing and polymorphism screen of 10 sooty mangabeys
DNA was prepared from blood or liver samples from 10 sooty mangabeys from the YNPRC colony. Ten sooty mangabey breeder animals were selected in consultation with the YNPRC Breeding Manager representing at least 90% of colony diversity based on the pedigree of the colony. Illumina paired-end libraries (300-bp insert size) were prepared as described above for 500-bp paired-end libraries. These libraries were sequenced (100 bp reads) on a HiSeq2000 instrument, producing an average of 30× whole-genome coverage across individuals. These reads were mapped to the C. atys assembly using BWA-mem and single-nucleotide variants were called using GATK (https://software.broadinstitute.org/gatk/). A gVCF file was created for each animal, and variation in the regions of interest for TLR4 and ICAM2 were identified in those files.
Polymorphism screen among rhesus macaques
To assess variation in TRL4 and ICAM2 among rhesus macaques, we used our database of whole-genome sequence data from 133 individuals of this species. The details of sequencing and single-nucleotide variants discovery for this population have previously been described8. The population-level VCF file for this study was examined for relevant variation in these two genes.
Targeted re-sequencing of ICAM2 and TLR4 in rhesus macaques and sooty mangabeys
To test the validity of the apparent species differences in ICAM2 and TLR4 between rhesus macaques and sooty mangabeys, primers were designed to flank three areas of interest (see Extended Data Figs 3a, 5b), PCR was performed using genomic DNA from two rhesus macaques and two sooty mangabeys (including FAK, the animal used for the Caty_1.0 reference genome) and the PCR product was subjected to Sanger sequencing. PCR primers were designed using Primer3 with default settings with the exception that the human mis-priming library was selected (http://bioinfo.ut.ee/primer3/)28,29. Primers were tailed with M13 sequences to facilitate Sanger sequencing.
PCR primer pairs (gene specific sequences are underlined): ICAM2_Ex2_F GTAAAACGACGGCCAGTATGTGCAGGTGGAGTGTGAT; ICAM2_Ex2_R GGAAACAGCTATGACCATGGCTCGAACAGACTCAGTGGA; ICAM2_Ex3_F GTAAAACGACGGCCAGTAAGCAGAGCAGGACAGATGT; ICAM2_Ex3_R GGAAACAGCTATGACCATGACTCTGCACAGTCAGACCTT; TLR4_SL_F GTAAAACGACGGCCAGTACCATGGAATGACTTGCCCT; TLR4_SL_R GGAAACAGCTATGACCATGCCTTTCAGCTCTGCCTTCAC.
AmpliTaq Gold 360 DNA Polymerase (Applied Biosystems) was used to amplify PCR products using the following protocol: 95 °C for 10 min; 95 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s, 10 cycles (annealing temperature is decreased by 1 °C per cycle); 94 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, 30 cycles; 72 °C for 10 min. PCR products were subjected to Sanger sequencing (in both directions) using M13 primers. PCR and Sanger sequencing was performed at ACGT. Traces (see Fig. 2a for examples) were inspected and consensus sequences obtained for each PCR product. Primer sequences were trimmed and consensus sequences were deposited in GenBank (accession numbers: MF468275–MF468286).
Sequencing and de novo assembly of RNA-seq transcripts
Transcripts for sooty mangabey were assembled de novo from RNA-seq reads using Trinity on XSEDE’s Blacklight supercomputer30. The RNA-seq reads were pooled from 12 different tissues and were prepared by the standard mRNA-seq with the uracil DNA glycosylase protocol (Illumina kit Part RS-122-2303) and are publicly available from the Nonhuman Primate Reference Transcriptome Resource (NCBI SRA accession numbers SRX270666 and SRX270667)27. We performed a number of filtering steps to prepare threads for de novo assembly, which included removing adapters, filtering for quality, removing poly A/T tails and removing mtDNA and common mammalian rRNA27,31. After filtering, we used an input of 1,635,074,685 RNA-seq reads as the basis for the transcriptome assembly. Using around 550 mostly continuous compute hours on Blacklight, we partitioned the computational job into three phases described by the Trinity algorithm: Inchworm (around 100 h × 64 cores), Chrysalis (around 400 h × 128 cores), and Quantify Graph and Butterfly (around 50 h × 64 cores). To circumvent the large amount of I/O generated in the Quantify Graph phase, we ran Trinity directly from the RAM disk for this phase. Using Trinity (version r2012-10-05), the following options were selected:
Trinity.pl–JM 512G–no_run_chrysalis–seqType fa–single, reads.fasta–run_as_paired–CPU 16, Trinity.pl–JM 512G–no_run_quantifygraph–seqType fa–single, reads.fasta–run_as_paired–CPU 16–bflyGCThreads 4, Trinity.pl–JM 512G–no_run_butterfly–seqType fa–single
reads.fasta–run_as_paired–CPU 16., Trinity.pl–JM 512G–bflyGCThreads 16–bflyCPU 32–seqType fa, –single reads.fasta–run_as_paired–CPU 16.
The large N25 (6,431 bp), N50 (3,483 bp) and N75 (1,116bp) values of the resulting assembly were indicative of its success.
Pipeline for finding divergent sooty mangabey proteins
C. atys assembly Caty_1.0 protein model predictions were screened against the curated M. mulatta MacaM protein models by alignment with BLASTp (v.2.2.28+)22. The C. atys protein model alignment with the lowest e value or highest bitscore (for equal e values) was selected for each MacaM protein model, yielding the set of orthologous C. atys protein predictions most similar to the M. mulatta protein models. The spliced CDS sequence for each Caty_1.0 transcript prediction was extracted with gffread (utility from cufflinks v.2.1.1). Caty_1.0 transcript prediction CDS sequences were screened against the de novo RNA-seq assembly transcript models by alignment with BLAT (v.34) and an aligment score was calculated as the number of matching bases minus the number of CDS sequence bases missing in alignment gaps normalized by the CDS sequence length.
This score penalizes bases missing from the CDS sequence without penalizing extra sequence that may have been added to the RNA-seq transcript model during the assembly process. Only predicted CDS sequences that had a score >0.99 were retained as supported by RNA-seq data. The MacaM best match selected Caty1.0 protein models were then cross-referenced with the RNA-seq supported Caty_1.0 transcript models to eliminate protein models without RNA-seq evidence. The protein alignments to MacaM for these models were then re-examined to find genes for which the alignment identity was less than 97%, where there were gaps in the alignments or the alignment was not the full length of the protein model. These two species share a common ancestor about 10–11 million years ago, and therefore the expectation is that most proteins will be >97% identical. This was confirmed by using a maximum likelihood amino acid model (WAG amino acid matrix) to estimate sequence distances between the C. atys and M. mulatta orthologues (Extended Data Fig. 1). Proteins of interest for differential response to lentivirus infection may be more divergent than expected on average. These represent potentially divergent genes and were further screened against the Gene Ontology (GO) term ‘immune response’. This list of divergent immune genes was then further curated by manual inspection of multiple alignments of cDNA transcript and genomic sequences of C. atys (Caty_1.0), M. mulatta (MacaM) and human (GRCh38.p7). Multiple alignment analysis was performed using Multalin (http://multalin.toulouse.inra.fr/). TLR4 and ICAM2 sequence alignments were generated using Jalview.
Gene family evolution methods
In order to identify rapidly evolving gene families along the C. atys lineage, we obtained peptides from human, chimpanzee, orangutan, gibbon, macaque, baboon, vervet, marmoset and mouse from ENSEMBL 8332. The C. atys peptides were obtained from NCBI33. To ensure that each gene was counted only once, we used only the longest isoform of each protein in each species. We then performed an all-versus-all BLAST search on these filtered sequences34. The resulting e values from the search were used as the main clustering criterion for the MCL program to group peptides into gene families35. This resulted in 14,889 clusters. We then removed all clusters only present in a single species, resulting in 10,967 gene families. We also obtained an ultrametric tree from a previous study and added sooty mangabey based on its divergence time from baboon (TimeTree)36,37.
With the gene family data and ultrametric phylogeny as input, we estimated gene gain and loss rates (λ) with CAFE v.3.038. This version of CAFE is able to estimate the amount of assembly and annotation error (ε) present in the input data using a distribution across the observed gene family counts and a pseudo-likelihood search. CAFE is then able to correct for this error and obtain a more accurate estimate of λ. We find an ε of about 0.04, which implies that 4% of gene families have observed counts that are not equal to their true counts. After correcting for this error rate, we find λ = 0.0020. These values for ε and λ are on par with those previously reported for mammalian datasets38,39 (Extended Data Table 3b). Using the estimated λ value, CAFE infers ancestral gene counts and calculates P values across the tree for each family and lineage to assess the significance of any gene family changes along a given branch. CAFE uses Monte Carlo re-sampling to assess if a given family is rapidly evolving. For those families found to be rapidly evolving (P < 0.01), it then calculates P values for each lineage within the family using the Viterbi method. Those lineages with low P values (P < 0.01) are said to be rapidly evolving.
We observed 1,561 rapidly evolving families across the 10 species of mammals sampled here. Extended Data Table 3c summarizes the gene family changes for all 10 species. Humans have the highest average expansion rate across all families at 0.20 whereas gibbons have the lowest at −0.09, meaning that they have the most gene family contractions. C. atys has undergone 535 gene family expansions of which 96 are rapid expansions and 340 gene family contractions of which 48 are rapid contractions.
Genetic distance between C. atys and M. mulatta orthologues
The amino acid sequences of 9,257 C. atys proteins with RNA-seq support (Fig. 1) were aligned to M. mulatta orthologues as described above. We then used the codeml package from PAML (v.4.9a) on each of these alignments with the WAG amino acid rate matrix to calculate maximum likelihood genetic distances between the two sequences40. A histogram was generated from these distances with R (Extended Data Fig. 1a).
TLR4 gene tree
TLR4 nucleotide sequences for 17 primate species were obtained from the NCBI GenBank resource (human: NM_138554.4; rhesus macaque: XM_015116960.1; sooty mangabey: manually curated XM_012091593.1; bonobo: NM_001279223.1; Nancy Ma’s night monkey: XM_012472756.2; drill: XM_011973281.1; colobus monkey: XM_011950060.1; crab-eating macaque: NM_001319615.1; squirrel monkey: XM_003925187.2; baboon: XM_003911309.4; pig-tailed macaque: NM_001305889.1; marmoset: XM_017975811.1; gorilla: XM_004048514.2; chimpanzee: NM_001144863.1; orangutan: AB445642.1; African green monkey: XM_007968248.1; gibbon: XM_003264057.3). These sequences were aligned with PASTA2 and we then constructed a maximum likelihood gene tree with RAxML3, performing 100 bootstrap replicates41,42 (Extended Data Fig. 7). Finding low bootstrap support amongst nodes ancestral to sooty mangabey, drill and baboon, we counted the number of sites that were discordant with respect to the gene tree topology. That is, the number of sites in which baboon and C. atys share the same state and C. atys and drill share a different state with an outgroup species (one of the two other Old World monkeys).
Sample collection and processing
Peripheral blood samples from SIV-negative rhesus macaques and SIV-negative sooty mangabeys were collected by venipuncture according to standard procedures at the Yerkes National Primate Research Center of Emory University and in accordance with US National Institutes of Health guidelines. Human blood samples were obtained from healthy donors at the Yerkes National Primate Research Center in accordance with Institutional Review Board protocol IRB0004582 and all relevant ethical regulations. Informed consent was obtained from all blood donors. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using Ficoll density-gradient centrifugation.
In vitro TLR-ligand stimulation assay
The assay used in this study is a modified version of the procedure previously described43. Ultrapure LPS (Escherichia coli 0111:B4) and monophosphoryl lipid-A (Salmonella minnesota) were purchased from Invivogen. Whole blood collected in EDTA vacutainers was diluted 1:4 with RPMI 1640 medium and 195 μl aliquots were transferred to 96-well, round-bottom micro-titre plates. Agonists were diluted in RPMI 1640 and 5 μl were applied to the wells at the following final concentrations: LPS, 1,000–10 ng ml−1; lipid-A, 10–1 μg ml−1. Suspensions were then mixed by pipet and incubated at 37 ° C, 5% CO2 for 4 h). After incubation, plates were centrifuged at 700 r.p.m. for 10 min, and 120 μl of cell-free supernatant was removed and stored at −80 °C until the assay was carried out. Each TLR ligand at a given concentration was performed in triplicate for each animal.
Cytokine bead array (CBA)
Samples were obtained from sooty mangabeys and rhesus macaques housed at the YNPRC. Sooty mangabeys were naturally infected at the YNPRC and rhesus macaques had been infected previously with SIVsmm as previously described19. Supernatant levels of TNF and IL-6 were measured using the human inflammation CBA kit (BD Biosciences Immunocytometry Systems) according to the manufacturer’s instructions, with the modification that the sample volumes for supernatant, antibody-coupled bead mix and PE-conjugated detection antibody solution were all reduced to 25 μl instead of 50 μl44. After incubation, samples were washed with 2% paraformaldehyde in PBS, resuspended in 150 μl PBS, and analysed using a FACSCalibur flow cytometer (BD Biosciences Immunocytometry Systems). The average of triplicate cytokine measurements was used as the representative value for individual animals, and variations in cytokine levels between species groups were tested for statistical significance using unpaired t-tests in Prism 6.0. To quantify the level of TLR4 mRNA, and to perform linear regression of TLR-signalling molecules with TNF and IL6 cytokine levels, in the LPS-stimulated blood samples in the longitudinal SIVsmm-infected samples, we used microarray expression data from matched whole-blood samples; these data are available from the NCBI Geo database (accession GSE16147).
Plasma viral load measurement
Quantification of SIVsmm plasma viral RNA levels were quantified using qPCR as described previously45,46.
RNA-seq analysis of LPS-stimulated monocytes
RNA-seq analysis was conducted at the Yerkes Nonhuman Primate Genomics Core Laboratory (http://www.yerkes.emory.edu/nhp_genomics_core/). CD14+ monocytes were isolated from Ficoll-isolated PBMCs using CD14 MicroBeads according to the manufacturer’s instructions (Miltenyi Biotec). Subsequently, 0.4 × 106 cells were stimulated for 6 h with 10 ng ml−1 LPS and then immediately lysed in 350 μl RLT buffer (Qiagen). RNA was purified using Micro RNEasy columns (Qiagen) and RNA quality was assessed using Agilent Bioanalyzer. Then, 10 ng of total RNA was used as input for mRNA amplification using 5′ template-switch PCR with the Clontech SMART-Seq v.4 Ultra Low Input RNA kit, according to the manufacturer’s instructions. Amplified mRNA was fragmented and appended with dual indexed barcodes using Illumina NexteraXT DNA Library Prep kits. Libraries were validated by capillary electrophoresis on an Agilent 4200 TapeStation, pooled and sequenced on an Illumina HiSeq 3000 using (100 bp paired-end reads) at an average read depth of 18 million. RNA-seq data were analysed by alignment and annotation to either the MacaM v.7.8.2 assembly of the Indian rhesus macaque genome (available at https://www.unmc.edu/rhesusgenechip/index.htm) or to the Caty_1.0 assembly22. Alignment was performed using STAR v.2.5.2b using the annotation as a splice junction and abundance estimation reference, and non-unique mappings were removed from downstream analysis47. Transcripts were annotated using both the MacaM and Caty 1.0 assemblies and annotation as described in the text. Transcript abundance was estimated internally in STAR using the algorithm of HT-Seq and differential expression analyses were performed using the DESeq2 packages48,49. To quantitatively compare the degree to which LPS treatment induced inflammatory gene expression between species, we used GSEA50. GSEA was performed using the desktop module available from the Broad Institute (https://www.broadinstitute.org/gsea/)51. Gene ranks for contrasts of LPS-treated versus untreated samples were calculated from the normalized expression tables using the signal-to-noise metric for each species separately. Ranked datasets contrasting LPS-treated versus untreated samples were tested for enrichment of the gene sets ‘HALLMARK_TNFA_SIGNALING_VIA_NFKB’ (M5890) and ‘HALLMARK_IL6_JAK_ STAT3_SIGNALING’ (M5897) from the Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb/index.jsp) using gene set permutation to test for statistical significance. Heat maps and other visualizations were generated using Partek Genomics software, v.6.6.
ICAM2 exon splice junction analysis
RNA-seq alignments from all 24 LPS-stimulated monocyte samples, and alignments derived from deep RNA-seq (over 50 million reads) from two samples derived from flow-sorted, purified, blood C. atys conventional dendritic cells (cDCs, defined as CD3−CD14−CD20−CD123−HLA-DR+CD11c+) that were prepared alongside the monocytes were examined for observed splicing. To provide additional depth, we also included RNA-seq data from two flow-purified M. mulatta ‘non-classical’ monocyte samples (defined as CD14−CD16+HLA-DR+NKG2−CD3−CD20−) and one C. atys sample from CD4+ T transitional memory cells (CD4+ TTM, defined as CD3+CD4+CD8−CD45RA−CD95+CD28+CCR7highCD62L−CD14−CD16−CD20−). Reads from the alignment (BAM) files that mapped from 5 kb upstream to 5 kb downstream of the ICAM2 loci were scanned by a custom Perl script that recorded evidence of splicing from the CIGAR field, and accumulated counts of reads supporting either splicing or read-through at each site. Splice site counts for all the samples were added together and compared to find the proportion of reads supporting each splice variant or intronic retention.
NF-κB luciferase reporter assay
Protein expressing constructs encoding human TLR4, MmTLR4, CaTLR4, MmTLR4 with the C terminus of CaTLR4, and CaTLR4 with the C terminus of MmTLR4 were generated by the Emory Custom Cloning Core Division using standard cloning techniques. HEK293T were obtained from ATCC and regularly checked for mycoplasma contamination.
To determine the responsiveness of MmTLR-4 and CaTLR-4 to LPS, HEK293T cells were seeded in poly-l-lysine-coated 96-well plates and transfected in triplicate using a standard calcium phosphate transfection protocol. Cells were co-transfected with expression plasmids of human MD-2 (pEFBOS, 5 ng), human CD14 (pcDNA3, 5 ng) and different TLR-4 orthologues or chimaeras (pEF1a, 2.5 ng). The MD-2- and CD14-expression plasmids were provided by A. Medvedev; the NF-κB reporter construct was made available by B. Baumann52,53. A firefly-luciferase reporter under the control of three NF-κB-binding sites (100 ng) and a Gaussia luciferase reporter (5 ng) under the control of the pTAL promoter were co-transfected to monitor NF-κB activity. The pTAL promoter construct contains a minimal TATA-like promoter (pTAL) region from the herpes simplex virus thymidine kinase (HSV-TK) promoter (Clontech) that is nonresponsive to NF-κB and served as an internal control. To activate NF-κB, cells were stimulated with 5 μg ml−1 LPS (E. coli 026:B6, eBioscience) for 5 h. After 40 h of transfection, a dual luciferase assay was performed and the firefly luciferase signals were normalized to the corresponding Gaussia luciferase control values.
qPCR
TLR stimulations of whole blood for qPCR were performed using the same method as for cytokine protein assay but scaled proportionally to use 1 ml of blood as input. Following stimulation, leukocytes were recovered by centrifugation at 700 r.p.m. for 5 min and removal of erythrocytes by incubation in ACK lysis buffer. Cells were lysed in 350 μl of RLT buffer, and RNA purified using the RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. qPCR was performed on RNA as previously described54. Primers to cytokines for qPCR were designed using Primer Express software (Applied Biosystems) to regions of 100% nucleotide identity between M. mulatta and C. atys: 12S rRNA (endogenous standard) forward 5′-CCCCCTAGAGGAGCCTGTTC-3′, 12S rRNA reverse 5′-GGCGGTATATAGGCTGAGCAA-3′; TNF forward 5′-GCCCTGGTATGAGCCCATCTA-3′, TNF reverse 5′-CGAGATAGTCGGGCAGATTGA-3′; IL6 forward 5′-GAGAAAGGAGACATGTAACAGGAGTAAC-3′, IL6 reverse 5′-TGGAAGGTTCAGGTTGTTTTCTG-3′. Fold change was calculated by dividing the normalized post-treatment sample quantity with the normalized untreated control quantity from the same animal, and calculating the average of fold changes for each species.
Flow cytometry of PBMCs
Multicolour flow cytometry staining was performed using the following antibodies and reagents: CD3–APC/Cy7 (SP34-2), CD14–PE/Cy7 (M5E2) and CD20–PE/Cy5 (2H7) from BD; CD4–BV650 (OKT4), CD8–BV711 (RPA-T8), ICAM-2–FITC (CBR-IC2/2), Mouse IgG2a(κ)–FITC (MOPC-173) isotype control from Biolegend; Live/Dead Fixable Aqua from Thermo Fisher Scientific. Cells were stained for flow cytometry and data were acquired on an LSR II cytometer (BD) and analysed by FlowJo 10 software (TreeStar). Further analyses were performed using PRISM (GraphPad) and Excel (Microsoft Office 2011) software.
ICAM-2 western blot
PBMCs were lysed in RIPA buffer and equal amounts of cell lysate were boiled after addition of sample buffer including β-mercaptoethanol, resolved with a 4–15% SDS–PAGE (Bio-Rad), and proteins were transferred to an Immobilon-P PVDF membrane (Millipore). Afterwards membranes were blocked for 1 h in blocking buffer (Bio-Rad) and incubated overnight with polyclonal rabbit ICAM-2-specific antibody (Bethyl). After washing (PBS with 0.05% Tween-20), anti-rabbit HRP-conjugated secondary antibody was incubated for an additional 1 h, washed, and HRP activity was determined using the Super Signal West Pico Kit (Bio-Rad and visualized using the ChemiDoc XRS+ (Bio-Rad). Then the membrane was stripped with buffer (2% SDS, 0.5 M Tris, pH 2.2), blocked again and β-actin was detected using a rabbit anti-β-actin antibody as primary antibody and anti-rabbit-HRP antibody as secondary antibody.
Statistical analysis
Statistical significance was determined using an unpaired Student’s t-test with Welch’s correction. P < 0.05 was considered significant. *P < 0.05; **P < 0.01; NS, not significant. Data are mean ± s.d. or s.e.m. as indicated. Significance for comparisons of mRNA levels of individual genes in RNA-seq data was tested using the Wald test as part of the DESeq2 workflow. Bars represent group means, and dots represent read counts for individual samples normalized to library size. P values denoted are adjusted using Benjamini–Hochberg correction.
Code availability
We used a custom script to quantify ICAM-2 splice junctions. This script is available at Github: https://github.com/BosingerLab/splicing-analysis.
Data availability
Raw sequences of the C. atys reference genome have been deposited in NCBI under Bioproject accession number PRJNA157077. The genome assembly is available at NCBI as Caty1.0 (RefSeq accession GCF_000955945.1). The multi-tissue C. atys RNA-seq reads are available from the Nonhuman Primate Reference Transcriptome Resource (NCBI SRA accession numbers SRX270666 and SRX270667). Data from Sanger sequencing of TLR4 and ICAM2 are available at NCBI (accession numbers MF468275–MF468286). Microarray data used for TLR-4 measurement and linear regression with TNF and IL-6 are available from the NCBI GEO database (accession GSE16147). The RNA-seq data for LPS-stimulated monocytes was submitted to the GEO database (accession numbers GSM2711028–GSM2711051 and GSE101617).
Accession codes
Primary accessions
BioProject
GenBank/EMBL/DDBJ
Gene Expression Omnibus
Sequence Read Archive
References
Chahroudi, A., Bosinger, S. E., Vanderford, T. H., Paiardini, M. & Silvestri, G. Natural SIV hosts: showing AIDS the door. Science 335, 1188–1193 (2012)
Cartwright, E. K. et al. Divergent CD4+ T memory stem cell dynamics in pathogenic and nonpathogenic simian immunodeficiency virus infections. J. Immunol. 192, 4666–4673 (2014)
Pandrea, I. et al. Paucity of CD4+ CCR5+ T cells may prevent transmission of simian immunodeficiency virus in natural nonhuman primate hosts by breast-feeding. J. Virol. 82, 5501–5509 (2008)
Paiardini, M. et al. Low levels of SIV infection in sooty mangabey central memory CD4+ T cells are associated with limited CCR5 expression. Nat. Med. 17, 830–836 (2011)
Beaumier, C. M. et al. CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat. Med. 15, 879–885 (2009)
Halai, K., Whiteford, J., Ma, B., Nourshargh, S. & Woodfin, A. ICAM-2 facilitates luminal interactions between neutrophils and endothelial cells in vivo. J. Cell Sci. 127, 620–629 (2014)
Staunton, D. E., Dustin, M. L. & Springer, T. A. Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature 339, 61–64 (1989)
Xue, C. et al. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 26, 1651–1662 (2016)
Casasnovas, J. M., Pieroni, C. & Springer, T. A. Lymphocyte function-associated antigen-1 binding residues in intercellular adhesion molecule-2 (ICAM-2) and the integrin binding surface in the ICAM subfamily. Proc. Natl Acad. Sci. USA 96, 3017–3022 (1999)
Brenchley, J. M. et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12, 1365–1371 (2006)
Brenchley, J. M. & Douek, D. C. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 1, 23–30 (2008)
Perelman, P. et al. A molecular phylogeny of living primates. PLoS Genet. 7, e1001342 (2011)
Mendes, F. K. & Hahn, M. W. Gene tree discordance causes apparent substitution rate variation. Syst. Biol. 65, 711–721 (2016)
Finstermeier, K. et al. A mitogenomic phylogeny of living primates. PLoS ONE 8, e69504 (2013)
Mir, K. D. et al. Reduced Simian immunodeficiency virus replication in macrophages of sooty mangabeys is associated with increased expression of host restriction factors. J. Virol. 89, 10136–10144 (2015)
Klatt, N. R. et al. Availability of activated CD4+ T cells dictates the level of viremia in naturally SIV-infected sooty mangabeys. J. Clin. Invest. 118, 2039–2049 (2008)
Raetz, C. R. & Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 (2002)
Rogers, J. & Gibbs, R. A. Comparative primate genomics: emerging patterns of genome content and dynamics. Nat. Rev. Genet. 15, 347–359 (2014)
Bosinger, S. E. et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J. Clin. Invest. 119, 3556–3572 (2009)
Jacquelin, B. et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119, 3544–3555 (2009)
Bosinger, S. E. et al. Intact type I interferon production and IRF7 function in sooty mangabeys. PLoS Pathog. 9, e1003597 (2013)
Zimin, A. V. et al. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol. Direct 9, 20 (2014)
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011)
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012)
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015)
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016)
Pipes, L. et al. The non-human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Res. 41, D906–D914 (2013)
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012)
Koressaar, T. & Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291 (2007)
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011)
Peng, X. et al. Tissue-specific transcriptome sequencing analysis expands the non-human primate reference transcriptome resource (NHPRTR). Nucleic Acids Res. 43, D737–D742 (2015)
Flicek, P. et al. Ensembl 2014. Nucleic Acids Res. 42, D749–D755 (2014)
Geer, L. Y. et al. The NCBI BioSystems database. Nucleic Acids Res. 38, D492–D496 (2010)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002)
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006)
Warren, W. C. et al. The genome of the vervet (Chlorocebus aethiops sabaeus). Genome Res. 25, 1921–1933 (2015)
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013)
Carbone, L. et al. Gibbon genome and the fast karyotype evolution of small apes. Nature 513, 195–201 (2014)
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007)
Mirarab, S. et al. PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J. Comput. Biol. 22, 377–386 (2015)
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014)
Tapping, R. I., Akashi, S., Miyake, K., Godowski, P. J. & Tobias, P. S. Toll-like receptor 4, but not toll-like receptor 2, is a signaling receptor for Escherichia and Salmonella lipopolysaccharides. J. Immunol. 165, 5780–5787 (2000)
Morgan, E. et al. Cytometric bead array: a multiplexed assay platform with applications in various areas of biology. Clin. Immunol. 110, 252–266 (2004)
Gordon, S. N. et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J. Immunol. 179, 3026–3034 (2007)
Sumpter, B. et al. Correlates of preserved CD4+ T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: implications for AIDS pathogenesis. J. Immunol. 178, 1680–1691 (2007)
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013)
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015)
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014)
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005)
Subramanian, A., Kuehn, H., Gould, J., Tamayo, P. & Mesirov, J. P. GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics 23, 3251–3253 (2007)
Medvedev, A. E. & Vogel, S. N. Overexpression of CD14, TLR4, and MD-2 in HEK 293T cells does not prevent induction of in vitro endotoxin tolerance. J. Endotoxin Res. 9, 60–64 (2003)
Sauter, D. et al. Differential regulation of NF-κB-mediated proviral and antiviral host gene expression by primate lentiviral Nef and Vpu proteins. Cell Rep. 10, 586–599 (2015)
Bosinger, S. E. et al. Gene expression profiling of host response in models of acute HIV infection. J. Immunol. 173, 6858–6863 (2004)
Acknowledgements
We thank S. Ehnert and all animal care and veterinary staff at the Yerkes National Primate Research Center (YNPRC); B. Cervasi and K. Gill at the Emory University Flow Cytometry Core; M. T. Nega at the Emory CFAR Virology & Drug Discovery Core; R. Linsenmeyer for excellent technical assistance; and O. Laur and the Emory Custom Cloning Core Division. We are grateful for the sequence production and related activities at the Human Genome Sequencing Center at Baylor College of Medicine. The production teams involved were: sample tracking (S. Jhangiani, C. Kovar), library production (H. Doddapaneni, H. Chao, S. L. Lee, G. Weissenberger and M. Wang), Illumina sequencing (H. Dinh, G. Okwuonu and J. Santibanez), PacBio sequencing (V. Vee) and production informatics (M. Dahdouli, Z. Khan, J. G. Reid and D. Sexton). D. Rio Derios and S. C. Murali also contributed to the assembly of the genome sequence. This work was funded by HHS/National Institutes of Health (NIH) (R37 AI66998). Research reported in this publication was also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number P51 OD011132 (to the YNPRC). D.M.M., R.A.G., K.W. and J.R. were supported by NIH grant U54-HG006484-01. D.S. was supported by the Priority Program ‘Innate Sensing and Restriction of Retroviruses’ (SPP 1923) of the German Research Foundation (DFG). F.K. was funded by the Advanced ERC grant ‘Anti-Virome’ and the DFG-funded SFB 1279. G.W.C.T. and M.W.H. were supported by the Precision Health Initiative of Indiana University. B.H.H. was funded by NIH grant R37 AI050529. Comparative transcriptomics research was funded by NIH grant R24 OD010445.
Author information
Authors and Affiliations
Contributions
D.P. and S.E.B. designed and performed experiments and analysed data. S.E.B. and G.K.T. designed and performed bioinformatics analyses. F.K. and B.H.H. designed experiments. T.H.V., M.P. and A.C. contributed to the study design and data interpretation. R.B.N. performed custom annotation of macaque and mangabey genomes and Sanger sequencing. Z.P.J. collected samples and analysed data. Y.H. contributed to sequencing. A.E. contributed to genome assembly. M.R., D.M.M., and R.A.G. supervised and/or managed the sequencing of the C. atys genome. R.A.H. and Y.L. performed genome assembly tasks. R.A.D. performed RNA-seq sample processing and analysis. D.L.S. analysed TLR-4 functional data. K.W. and J.R. supervised the assembly and analysis of the genome. C.-B.S. and S.M.S. analysed and interpreted genetic data of ICAM-2. G.W.C.T. and M.W.H. analysed gene family evolution. N.B.P. collected samples and conducted RNA-seq experiments. L.P. and C.E.M. sequenced and assembled RNA-seq transcripts. D.S. designed and analysed TLR-4 experiments. J.R. conceived the study, designed experiments and analysed genomic data. G.S. conceived, designed and led the study. D.P., S.E.B., J.R. and G.S. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks M. Martin and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Genetic distances of C. atys and M. mulatta orthologues and protein sequence alignments of CD4 and CCR5.
a, Genetic distances of C. atys and M. mulatta orthologues. The dotted blue line represents a mean distance of 0.00755 expected substitutions, and the solid red line represents the 97th percentile. This percentile indicates that 8,979 out of 9,257 genes have a distance less than 0.0294. b, Pairwise alignment of CD4 and CCR5 protein sequences for C. atys and M. mulatta. Sequences were aligned using Jalview v.2.9.0.
Extended Data Figure 2 Sequence alignment of ICAM-2 protein and exon sequence analysis of ICAM2.
a, Pairwise alignment of predicted ICAM-2 protein models for sooty mangabey and rhesus macaque. Exon structure is highlighted based on human ICAM-2. Alignment was performed using Jalview v.2.9.0. b, The sequence of exon 3 of CaICAM2 was confirmed in 10 additional individuals. Sequencing reads were aligned to the C. atys reference genome and visualized using Integrative Genomics Viewer (IGV). The red arrow indicates the position of the 499-bp genomic deletion in C. atys.
Extended Data Figure 3 Predicted model of the ICAM2 gene structure and ICAM2 genome sequence alignments.
a, Predicted model of ICAM2 gene structure of M. mulatta and C. atys and the location of PCR primers for Sanger sequencing. Light blue, untranslated region; dark blue, CDS; red lines, intronic sequence; dotted line, exonic and intronic sequences present in human ICAM2 and MmICAM2 but not in CaICAM2; red box, the sequence that would be intronic in MmICAM2, but which is included in the exonic sequence of CaICAM2; light-purple box for CaICAM2 exon 4 represents the fact that the exon 4 sequence in MmICAM2 is present in CaICAM2 but is not included in the CaICAM2 CDS due to a stop codon in the CaICAM2 exon 3. Primer positions are indicated by arrows. Predicted PCR products are indicated by thick lines. Primers Ex3_F and Ex3_R were designed to amplify a region spanning a putative genomic deletion which includes the 3′ region of CaICAM2 exon 3 and intron 3. b, Alignment of ICAM2 genomic sequences. Sanger sequencing of 2 rhesus macaques and 2 sooty mangabeys (including the Caty_1.0 reference animal) was performed to confirm the ICAM2 genomic deletion specific to C. atys. Starting at MmICAM2 nucleotide position 3166, sequences were aligned using Jalview v.2.9.0. Dashed lines denote the deletion in C. atys. RM, rhesus macaque; SM, sooty mangabey.
Extended Data Figure 4 ICAM2 splice junction analysis in C. atys and M. mulatta by RNA-seq read alignment.
a, Quantification of observed splicing. Splice site counts for RNA-seq read alignments were added together and sites with more than 100 total reads were compared to find the proportion of reads supporting each splice variant or intronic retention. b, MmICAM2 splicing analysed by RNA-seq read alignment to the reference genome and visualized in IGV. c, CaICAM2 splicing analysed by RNA-seq read alignment to the reference genome and visualized in IGV.
Extended Data Figure 5 Sequence alignment of TLR-4 and the structure of the TLR4 gene.
a, Pairwise alignment of TLR-4 protein sequences for C. atys and M. mulatta. The sequence difference at the C terminus is highlighted in red. Sequences were aligned using Jalview v.2.9.0. b, TLR4 gene structure and location of PCR primers. Light blue, untranslated region; dark blue, CDS; red lines, intronic sequence. Primer positions are indicated by arrows. Predicted PCR product is indicated by thick line. Primers TLR4_F and TLR4_R were designed to amplify a region including a putative stop-loss mutation present in CaTLR4 but not in MmTLR4. c, Chromatograms showing stop-loss (indicated by arrows) in the TLR4 gene in C. atys with respect to M. mulatta. The relevant codon is underlined.
Extended Data Figure 6 TLR-4 C terminus sequence aligments.
a, The sequence of the CaTLR-4 C terminus was confirmed in 10 additional individuals. Sequencing reads were aligned to the Caty_1.0 reference genome and visualized in IGV. The red arrow indicates the position of the G-to-A stop codon mutation that can be found in MmTLR4 but not CaTLR4. b, Alignment of genomic sequences encoding the TLR4 C terminus from different primate species. Starting at human TLR4 nucleotide position 2461, sequences were aligned using Jalview v.2.9.0.
Extended Data Figure 7 Maximum likelihood gene tree of TLR4.
This topology corresponds to the accepted species relationships for Old World monkeys. However, low bootstrap support among the nodes ancestral to C. atys, drill and baboon indicate that several sites within the gene do not support that ordering and may be indicative of incomplete lineage sorting. The table on the left shows these sites.
Extended Data Figure 8 Analysis of cytokine expression and release after activation of TLR-4.
a, TNF release from whole blood upon stimulation with lipid-A. Whole blood was stimulated with lipid-A at the indicated concentrations for 4 h and cytokine secretion was measured by cytometric bead array. n = 5 biologically independent samples for M. mulatta; n = 4 biologically independent samples for C. atys. b, IL-6 release from whole blood upon stimulation with lipid-A. Whole blood was stimulated with lipid-A at the indicated concentrations for 4 h and cytokine secretion was measured by cytometric bead array. n = 8 biologically independent samples for M. mulatta; n = 9 biologically independent samples for C. atys. c, SIVsmm plasma viral load for M. mulatta and C. atys. SIVsmm RNA levels in plasma were quantified at the indicated time points after intravenous inoculation with a primary uncloned SIVsmm C. atys isolate. n = 5 biologically independent samples for each species. d, TLR4 mRNA levels in LPS-stimulated blood samples. To test the level of TLR4 expression in the LPS-stimulated blood samples shown in Fig. 3e, we isolated RNA from whole blood from time-point matched replicate samples using PAXgene Blood RNA tubes, and analysed expression using Affymetrix GeneChip Rhesus Macaque Genome Arrays, which contains three independent probesets specific for MmTLR4 (denoted on the x axis). Probeset intensities are displayed along the y axis as RMA normalized values. n = 3 biologically independent samples for M. mulatta; n = 4 biologically independent samples for C. atys. a–d, Dots represent individual animals, and the bar represents the mean. Unpaired two-sided Student’s t-test, P values are indicated. e, TNF and IL6 mRNA levels in LPS-stimulated monocytes from M. mulatta and C. atys. RNA-seq was used to assay global changes in gene expression after LPS stimulation of primary CD14+ monocytes. Significance for comparisons of mRNA levels of individual genes was tested using the Wald test as part of the DESeq2 workflow. Bars represent group means, and dots represent read counts for individual samples normalized to library size. Indicated P values are adjusted using the Benjamini–Hochberg correction. n = 6 biologically independent samples for each species.
Extended Data Figure 9 LPS-mediated induction of TNF and IL-6 inflammatory signalling is globally attenuated in C. atys.
a, b, Data shown are the leading-edge genes depicted in Fig. 3f, g (GSEA plots), for TNF-signalling genes (a) and IL-6-signalling genes (b). Values are the log2-transformed difference between LPS-treated and untreated samples for each individual animal. Genes selected are the combination of leading-edge/core-enriched genes for M. mulatta and C. atys GSEA analyses for each pathway. The gene sets selected for enrichment testing were obtained from the MSIGDB database hallmark collection are denoted at the top of each panel. Genes were organized using hierarchical clustering with Spearman dissimilarity and average linkage to estimate distance between genes and clusters, respectively. The colour scale at the bottom denotes the maximum and minimum on a log2 scale. For animal study source data, see Supplementary Table 2.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-3 and Supplementary Tables 1-2. (PDF 901 kb)
Rights and permissions
The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palesch, D., Bosinger, S., Tharp, G. et al. Sooty mangabey genome sequence provides insight into AIDS resistance in a natural SIV host. Nature 553, 77–81 (2018). https://doi.org/10.1038/nature25140
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25140
This article is cited by
-
Pro-inflammatory feedback loops define immune responses to pathogenic Lentivirus infection
Genome Medicine (2024)
-
Reduction of bitter taste receptor gene family in folivorous colobine primates relative to omnivorous cercopithecine primates
Primates (2024)
-
Current advances in primate genomics: novel approaches for understanding evolution and disease
Nature Reviews Genetics (2023)
-
Heterogeneity of circulating CD4+CD8+ double-positive T cells characterized by scRNA-seq analysis and trajectory inference
Scientific Reports (2022)
-
Genomic resources for rhesus macaques (Macaca mulatta)
Mammalian Genome (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
