
Abstract
Seahorses have a specialized morphology that includes a toothless tubular mouth, a body covered with bony plates, a male brood pouch, and the absence of caudal and pelvic fins. Here we report the sequencing and de novo assembly of the genome of the tiger tail seahorse, Hippocampus comes. Comparative genomic analysis identifies higher protein and nucleotide evolutionary rates in H. comes compared with other teleost fish genomes. We identified an astacin metalloprotease gene family that has undergone expansion and is highly expressed in the male brood pouch. We also find that the H. comes genome lacks enamel matrix protein-coding proline/glutamine-rich secretory calcium-binding phosphoprotein genes, which might have led to the loss of mineralized teeth. tbx4, a regulator of hindlimb development, is also not found in H. comes genome. Knockout of tbx4 in zebrafish showed a ‘pelvic fin-loss’ phenotype similar to that of seahorses.
Similar content being viewed by others
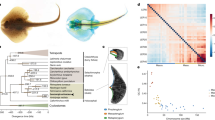

Main
Members of the teleost family Syngnathidae (seahorses, pipefishes and seadragons) (Extended Data Fig. 1), comprising approximately 300 species, display a complex array of morphological innovations and reproductive behaviours. This includes specialized morphological phenotypes such as an elongated snout with a small terminal mouth, fused jaws, absent pelvic and caudal fins, and an extended body covered with an armour of bony plates instead of scales1 (Fig. 1a). Syngnathids are also unique among vertebrates due to their ‘male pregnancy’, whereby males nourish developing embryos in a brood pouch until hatching and parturition occurs2,3. In addition, members of the subfamily Hippocampinae (seahorses) exhibit other derived features such as the lack of a caudal fin, a characteristic prehensile tail, and a vertical body axis4 (Fig. 1a). To understand the genetic basis of the specialized morphology and reproductive system of seahorses, we sequenced the genome of the tiger tail seahorse, H. comes, and carried out comparative genomic analyses with the genome sequences of other ray-finned fishes (Actinopterygii).

a, Schematic diagram of a pregnant male seahorse. b, The phylogenetic tree generated using protein sequences. The values on the branches are the distances (number of substitutions per site) between each of the teleost fishes and the spotted gar (outgroup). Spotted gar, Lepisosteus oculatus; zebrafish, Danio rerio.
Genome assembly and annotation
The genome of a male H. comes individual was sequenced using the Illumina HiSeq 2000 platform. After filtering low-quality and duplicate reads, 132.13 Gb (approximately 190-fold coverage of the estimated 695 Mb genome) of reads from libraries with insert sizes ranging from 170 bp to 20 kb were retained for assembly. The filtered reads were assembled using SOAPdenovo (version 2.04) to yield a 501.6 Mb assembly with an N50 contig size and N50 scaffold size of 34.7 kb and 1.8 Mb, respectively. Total RNA from combined soft tissues of H. comes was sequenced using RNA-sequencing (RNA-seq) and assembled de novo. The H. comes genome assembly is of high quality, as >99% of the de novo assembled transcripts (76,757 out of 77,040) could be mapped to the assembly; and 243 out of 248 core eukaryotic genes mapping approach (CEGMA) genes are complete in the assembly.
We predicted 23,458 genes in the genome of H. comes based on homology and by mapping the RNA-seq data of H. comes and a closely related species, the lined seahorse, Hippocampus erectus, to the genome assembly (see Methods and Supplementary Information). More than 97% of the predicted genes (22,941 genes) either have homologues in public databases (Swissprot, Trembl and the Kyoto Encyclopedia of Genes and Genomes (KEGG)) or are supported by assembled RNA-seq transcripts. Analysis of gene family evolution using a maximum likelihood framework identified an expansion of 25 gene families (261 genes; 1.11%) and contraction of 54 families (96 genes; 0.41%) in the H. comes lineage (Extended Data Fig. 2 and Supplementary Tables 4.1, 4.2). Transposable elements comprise around 24.8% (124.5 Mb) of the H. comes genome, with class II DNA transposons being the most abundant class (9%; 45 Mb). Only one wave of transposable element expansion was identified, with no evidence for a recent transposable element burst (Kimura divergence ≤ 5) (Extended Data Fig. 3).
Phylogenomics and evolutionary rate
The phylogenetic relationships between H. comes and other teleosts were determined using a genome-wide set of 4,122 one-to-one orthologous genes (Supplementary Note 4.2). The phylogenetic analysis (Fig. 1b) showed that H. comes is a sister group to other percomorph fishes analysed (stickleback, Gasterosteus aculeatus; medaka, Oryzias latipes; Nile tilapia, Oreochromis niloticus; fugu, Takifugu rubripes; and platyfish, Xiphophorus maculatus) with the exception of blue-spotted mudskipper (Boleophthalmus pectinirostris), a member of the family Gobiidae. Our inference, which placed the mudskipper as the outgroup, differs from that of a previous phylogenetic analysis based on fewer protein-coding genes that had placed syngnathids as an outgroup5. Estimated divergence times of H. comes and other teleosts calculated using MCMCTree suggest that H. comes diverged from the other percomorphs approximately 103.8 million years ago, during the Cretaceous period (Extended Data Fig. 2). Interestingly, the branch length of H. comes is longer than that of other teleosts, suggesting a higher protein evolutionary rate compared to other teleosts analysed in this study (Fig. 1b). This result was found to be statistically significant by both relative rate test6 and two cluster analysis7 (Supplementary Tables 4.3 and 4.4). To determine whether the neutral nucleotide substitution rate of H. comes is also higher, we generated a neutral tree on the basis of fourfold degenerate sites and calculated the pairwise distance of each teleost to the spotted gar (an outgroup) (Supplementary Fig. 4.4). The pairwise distance of H. comes was again higher compared with other teleosts, indicating that the neutral evolutionary rate of H. comes is also higher than that of other teleosts. The reasons for this higher molecular evolutionary rate in H. comes are unclear.
Gene loss
Gene loss or loss of function can contribute to evolutionary novelties and can be positively selected for8,9. We identified several genes that are not found in the H. comes genome but are found in other sequenced teleost genomes.
Secretory calcium-binding phosphoprotein (SCPP) genes encode extracellular matrix proteins that are involved in the formation of mineralized tissues such as bone, dentin, enamel and enameloid. Bony vertebrate genomes encode multiple SCPP genes that can be divided into two groups, the acidic and the proline/glutamine (P/Q)-rich SCPP genes. Acidic SCPPs regulate the mineralization of collagen scaffolds in bone and dentin whereas the P/Q-rich SCPPs are primarily involved in enamel or enameloid formation10. Analysis of the H. comes genome and the transcriptomes of H. comes and H. erectus showed that both contain two acidic SCPP genes, scpp1 and spp1 (Extended Data Fig. 4). However, no intact P/Q-rich gene could be identified. The only P/Q-rich gene present in the H. comes genome assembly, scpp5, is represented by only three out of ten exons, indicating that it has become a pseudogene. Seahorses and pipefish (family Syngnathidae) are toothless, a phenomenon known as edentulism. Besides syngnathids, edentulism has occurred convergently in several other vertebrate lineages11, the most notable ones being birds12, turtles, and some mammals such as baleen whales, pangolins and anteaters13. The loss of teeth in birds, turtles and mammals has been attributed to inactivating mutations in one or more P/Q-rich enamel-specific SCPP genes such as Enam, Amel, Ambn and Amtn, and the dentin-specific gene, Dspp12,14. In the case of H. comes, the complete loss of functional P/Q-rich SCPP genes may explain the loss of mineralized teeth.
Animals use their sense of smell, or olfaction, for finding food, mates and avoiding predators. Olfaction is mediated by olfactory receptors (ORs), which constitute the largest family of G-protein-coupled receptors. We were able to identify in the H. comes genome a significantly smaller repertoire of OR genes than in other teleosts (P value < 0.05, Wilcoxon rank-sum test). Our sensitive search pipeline (based on TblastN and Genewise) and manual inspection identified only 26 OR genes in the H. comes genome—the smallest OR repertoire identified in any ray-finned fish genome analysed so far (60 to 169 OR genes) (Fig. 2 and Extended Data Fig. 5).

‘Air’ and ‘water’ refer to the detection of airborne and water-soluble odorants, respectively. The sizes of the orange circles represent the number of OR genes of a particular category.
A derived phenotype of seahorse and other syngnathids is the complete lack of pelvic fins15,16. Pelvic fins are homologous to tetrapod hindlimbs and primarily serve a role in body trim and subtle swimming manoeuvres during teleost locomotion17,18,19. In addition, pelvic spines have an important role in protection against predators15. Pelvic fin loss has occurred independently in several teleost lineages, including Tetraodontidae (for example, pufferfishes), Anguillidae (eels) and Gasterosteidae (some populations of sticklebacks), and is frequently associated with a reduced pressure from predators and/or the evolution of an elongated body plan15. In pufferfish (fugu), pelvic fin loss is associated with a change in the expression pattern of hoxd9a20. In freshwater populations of stickleback, the loss of pelvic fins has been demonstrated to be due to deletions in the pelvic fin-specific enhancer of pitx1 (ref. 21).
Analysis of the H. comes genome and the transcriptomes of H. comes and H. erectus (see Supplementary Information, section 2), suggested that tbx4, a transcription factor conserved in jawed vertebrates, is not present in the seahorse genome (Fig. 3a) (Supplementary Information, section 9). To verify this, we carried out degenerate polymerase chain reaction (PCR) using genomic DNA from H. comes and several other species of syngnathids and some non-syngnathids. While the degenerate primers amplified a fragment of tbx4 from non-syngnathids, they failed to amplify a tbx4 fragment from syngnathid fishes (see Supplementary Information, section 9). Tbx4 is a T-box DNA-binding domain-containing transcription factor that acts as a regulator of hindlimb formation in mammals22,23,24. Loss of function of this gene in mouse leads to a failure of hindlimb formation22,23 as well as strong pleiotropic defects in lung25 and placental development22. Expression of zebrafish tbx4 specifically in pelvic fins suggests a similar role in appendage patterning in fishes24. Given the major role of tbx4 in hindlimb formation in mammals, we hypothesized that its absence in H. comes might be associated with the loss of pelvic fins. To test this hypothesis, we generated a CRISPR–Cas9 tbx4-knockout mutant zebrafish line. Interestingly, unlike homozygous mouse Tbx4 mutants, which fail to develop a functional allantois22, the homozygous zebrafish mutants are viable but completely lack pelvic fins without exhibiting any other gross morphological abnormalities in pectoral or median fins (Fig. 3c and Extended Data Fig. 6; see also Supplementary Information, section 9.3, in particular Supplementary Fig. 9.6 for additional phenotype analysis). This finding is consistent with the results of a recent study that showed that mutations in tbx4 are associated with the loss of pelvic fins in a naturally occurring zebrafish strain called pelvic finless26 (see also Supplementary Information, section 9.3). These results show that tbx4 has a role in pelvic fin formation in teleosts and suggests that the loss of pelvic fins in H. comes may be related to the loss of tbx4.

a, Vista plot of conserved elements in the tbx2b-tbx4-brip1 syntenic region in fugu (reference genome), seahorse (H. comes), stickleback and zebrafish showing that tbx4 is missing from this locus in seahorse. The blue and red peaks represent conserved exonic and non-coding sequences, respectively. b, Lateral (top) and ventral view (bottom) of wild-type (WT) and a representative (one out of five) F3 homozygous tbx4-null mutant (tbx4−/−) zebrafish. Bottom panel shows a close-up of the pelvic region (dashed lines indicate the approximate zoom region). Scale bar, 1 mm. Pelvic fins are indicated with black or white arrowheads in the wild-type fish. Homozygous tbx4-null mutants entirely lack pelvic fins without showing any other gross morphological defects.
Expansion of the patristacin gene family
Male pregnancy is an evolutionary innovation unique to syngnathids. In teleosts, the C6AST subfamily of astacin metalloproteases—such as high choriolytic enzyme (HCE) and low choriolytic enzyme (LCE)—are involved in lysing the chorion surrounding the egg, leading to hatching of embryos27. A member of this subfamily, patristacin (pastn), was found to be highly expressed in the brood pouch of pregnant males of the Gulf pipefish, Syngnathus scovelli, leading to the suggestion that this gene may have a role in the evolution of male pregnancy28. A pastn gene was also found to be highly expressed in the brood pouch of the male big belly seahorse, H. abdominalis, during mid- and late pregnancy29, suggesting a shared role for this gene in male pregnancy in syngnathids.
The H. comes genome contains six pastn genes (pastn1 to pastn6; Fig. 4a) organized in a cluster. To examine their expression patterns in the brood pouch, we carried out RNA-seq analysis at different stages of brood pouch development (see Supplementary Information, section 2) in H. erectus, as this species is easy to obtain and breed in the laboratory. H. comes and H. erectus exhibit very similar reproductive cycles and their coding sequences are highly similar (average identity of 93.3%; determined by aligning H. erectus RNA-seq transcripts to the H. comes genome assembly). We identified orthologues for five of the H. comes pastn genes (pastn1, pastn2, pastn3, pastn5 and pastn6) in the RNA-seq transcripts of H. erectus (Supplementary Fig. 2). Quantitative reverse transcription PCR (qRT–PCR) analysis of these genes showed that some of them are expressed at significantly higher levels in early- and late-pregnant stages (Fig. 4c). For example, pastn2 is expressed at significantly higher levels in early- and late-pregnant stages compared to the non-pregnant stage, whereas pastn1 and pastn3 are expressed at significantly higher levels during the late-pregnant stage compared to non-pregnant stage (Fig. 4c). This expression pattern suggests a role for these pastn genes in brood pouch development and/or hatching of embryos within the brood pouch prior to parturition.

a, Astacin gene loci in various ray-finned fish genomes showing expansion of pastn genes in seahorse (H. comes) and c6ast genes in platyfish. Chr, chromosome. b, The phylogeny of the astacin gene family in ray-finned fishes. Only pastn or c6ast genes shown in a are labelled. Supplementary Fig. 10.1 shows an expanded version of the tree with all the genes labelled. c, Expression patterns of pastn genes in relation to 18S ribosomal RNA genes in the brood pouch of male H. erectus determined by qRT–PCR. All data are expressed as mean ± standard error of mean (n = 5) and evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference test for adjusting P values from multiple comparisons (see Methods and Supplementary Information for details of methods). The average duration of pregnancy (from fertilization to parturition) is 17 days41. The y axis represents expression level in relation to 18S rRNA genes. pastn1 is expressed at low levels at the non-pregnant stage, which is not clearly visible in the figure due to the large scale used. Non-pregnant: no embryos in the brood pouch; early pregnant: 2–4 days post-fertilization; late pregnant: 12–14 days post-fertilization. *P < 0.05, **P < 0.01. Note that pastn4 is not expressed in these stages of brood pouch.
Interestingly, the platyfish (X. maculatus), in which fertilization and hatching of eggs occur within the maternal body (ovoviviparity), contains a cluster of six c6ast genes (Fig. 4a), with potential hatching enzyme-like activity30. Phylogenetic analysis of c6ast family genes in H. comes, platyfish and other fishes showed that H. comes pastn genes and platyfish c6ast genes form separate clades (Fig. 4b), indicating that they have expanded independently in the two lineages. Thus, this is an interesting instance of a gene family (C6AST subfamily of astacin metalloproteases) that has undergone expansion independently in different teleost lineages and shows new expression patterns and functions associated with similar evolutionary innovations (that is, ovoviviparity in female platyfish and male pregnancy in seahorse).
Loss of conserved noncoding elements
Vertebrate genomes contain thousands of noncoding elements that are under purifying selection31,32,33. Many of these conserved noncoding elements (CNEs) function as cis-regulatory elements such as enhancers, repressors and insulators34,35. Evolutionary loss of CNEs has important roles in phenotypic differences and morphological innovations21,36,37. To determine the extent of loss of CNEs in seahorse, we predicted genome-wide CNEs in H. comes and four other percomorph fishes (stickleback, fugu, medaka and Nile tilapia) using zebrafish as the reference genome (see Supplementary Information). We identified 239,976 CNEs (average size of 168 bp) that are conserved in zebrafish and at least one of the five percomorph fishes (Supplementary Table 6.1). To determine the extent to which CNEs are lost in H. comes, we searched for CNEs that are uniquely lost in each of the percomorph fishes. We restricted our analyses to a high-confidence set of CNEs situated in gap-free syntenic intervals (Supplementary Table 6.5). Interestingly, H. comes was found to have lost a substantially higher number of CNEs (1,612 CNEs) compared to other percomorphs (fugu, 1,050 CNEs; stickleback, 843 CNEs; medaka, 335 CNEs; Nile tilapia, 281 CNEs) (Supplementary Table 6.6).
Analysis of zebrafish CNEs that are lost in H. comes indicated that they are present in the neighbourhood of 728 genes enriched in functions such as regulation of transcription, regulation of the fibroblast growth factor receptor signalling pathway, embryonic pectoral fin morphogenesis, steroid hormone receptor activity and O-acetyltransferase activity (Supplementary Tables 6.8 and 6.9). The top 20 genes adjacent to regions with the highest number of CNEs lost in H. comes include sall1a, shox and irx5a (Supplementary Tables 6.10 and 6.11), which are involved in the development of the limbs, nervous system, kidney, heart and skeletal system. Altered expression patterns of these genes can potentially lead to altered morphological phenotypes. For example, loss of regulatory regions of the human SHOX gene is the cause of Leri–Weill dyschondrosteosis, a dominantly inherited skeletal dysplasia that is characterized by moderate short stature caused by short mesomelic limb segments38,39.
To verify the potential cis-regulatory functions of CNEs that were absent in H. comes but present in other teleost genomes, we assayed the function of seven selected zebrafish CNEs that were uniquely absent in H. comes. Of the seven CNEs assayed in transgenic zebrafish, four CNEs drove reproducible patterns of reporter gene expression in F1 embryos (Extended Data Fig. 7 and Supplementary Table 6.12). Thus, our transgenic assay indicates that some of the CNEs absent in H. comes may function as cis-regulatory elements in other teleosts. Further studies are required to examine whether the loss of CNEs may have played a role in the evolution of seahorse morphology.
Summary
Seahorses possess one of the most highly specialized morphologies and reproductive behaviours. We sequenced the genome of the tiger tail seahorse and performed comparative analysis with other teleost fishes. Our genome-wide analysis highlights several aspects that may have contributed to the highly specialized body plan and male pregnancy of seahorses. These include a higher protein and nucleotide evolutionary rate, loss of genes and expansion of gene families, with duplicated genes exhibiting new expression patterns, and loss of a selection of potential cis-regulatory elements. It is becoming recognized that evolutionary changes in cis-regulatory elements, particularly the loss and gain of enhancers, might play a major part in the evolution of morphological innovations and phenotypic changes across species21,36,37,40.
Male pregnancy is a unique developmental feature of seahorses and pipefishes (family Syngnathidae, comprising 57 genera and approximately 300 species). In the seahorse genome, the astacin subfamily of c6ast metalloprotease genes has undergone tandem duplications giving rise to six genes. This subfamily of metalloprotease includes the hatching enzyme (also known as choriolysin), HCE-like and HCE2-like enzymes that are responsible for hatching of embryos in fishes27. Of the six duplicated genes in seahorse, five are highly expressed in the male brood pouch, suggesting that they may be involved in male pregnancy, possibly through rewiring of their regulatory network. The loss of pelvic fins in seahorse is associated with the evolution of an armour-like covering of its body and gain of an elongated, flexible, substrate-gripping tail. By combining comparative genomics and gene-knockout experiments in zebrafish, we suggest that loss of tbx4 may have a role in this phenotype in seahorse. The loss of mineralized teeth in seahorse is associated with the fusion of the jaws into a tube-like snout and a small mouth, which is extremely efficient in sucking small food items that are abundant in the benthic environment. In teleosts, P/Q-rich SCPP genes are involved in the mineralization of enameloid, which is the equivalent of enamel in tetrapods10. The seahorse genome does not contain any intact P/Q-rich SCPP genes that code for enamel matrix proteins, suggesting that the loss of these genes could have played a part in the loss of its mineralized teeth. Our analyses of the H. comes genome sequence and comparative genomics with other teleosts highlighted several genetic changes that may be involved in the evolution of the unique morphology of seahorses.
Methods
Genome sequencing and assembly
Genomic DNA of a single male H. comes was used to construct eleven libraries including short-insert (170 bp, 500 bp, 800 bp) and mate-paired (2 kb, 5 kb, 10 kb, 20 kb) libraries and sequenced on the Illumina HiSeq 2000 sequencing platform. In total, we obtained around 218 Gb of raw sequence data (Supplementary Table 1.1). The genome was assembled using SOAPdenovo2.04 (ref. 42) with default parameters. No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
RNA sequencing and analysis
In total, 19 RNA-seq libraries were constructed, including two libraries from combined soft tissues (brain, gills, intestine, liver and muscle) from a male and a female H. comes (Supplementary Table 2.1); and 17 libraries of five developmental stages of embryos and different stages of brood pouch development such as the juvenile stage, rudimentary stage, pre-pregnancy stage, pregnancy stage, and post pregnancy stage, using RNA from the lined seahorse (Hippocampus erectus) (Supplementary Information, section 2). All libraries were prepared using Illumina TruSeq RNA sample preparation kit according to the manufacturer’s instructions (Illumina, San Diego, CA, USA) and sequenced using Illumina HiSeq 2000 platform. The RNA-seq reads were either de novo assembled using Trinity43 or mapped to the H. comes genome using TopHat44 with default parameters, and subsequently analysed using in-house Perl scripts. The differential expression of genes at different stages of brood pouch development was determined using the method developed previously45. The RNA-seq results were validated using qRT–PCR, with five biological replicates for each stage. All data were expressed as mean ± standard error of mean and were evaluated by one-way ANOVA followed by Tukey’s honestly significant difference test for adjusting P values from multiple comparisons. Results were considered to be statistically significant for P values < 0.05.
Genome annotation
Annotation of the H. comes genome was carried out using the Ensembl gene annotation pipeline which integrated ab initio gene predictions and evidence-based gene models. Briefly, protein sequences of D. rerio, G. aculeatus, O. latipes, T. rubripes and T. nigroviridis were downloaded from Ensembl (release 75) and mapped to the genome using TblastN46 with the parameter “-evalue 1E-5”. Second, high scoring segment pairs (HSPs) from blast were concatenated using Solar (in-house software, version 0.9.6). Third, the concatenated segments were aligned using GeneWise47 to refine the gene models. Finally, we filtered the alignments that showed alignment rates less than 50% of the full-length copies and filtered redundant alignments based on the GeneWise score. In addition, H. comes transcripts (female_transcript and male_transcript) and H. erectus transcripts (Juv_brain, Juv_body, Rud_testis and PreP_pouch) were used to assist in the gene model prediction. We annotated the predicted gene models using Swiss-Prot, TrEMBL, NCBI NR database, and KEGG databases (Supplementary Table 3.4).
Expansion and contraction of gene families
We used CAFE (version 2.1), a program for analysing gene family expansion and contraction under maximum likelihood framework. The gene family results from TreeFam pipeline and the estimated divergence time between species were used as inputs. We used the parameters “-p 0.01, -r 10000, -s” to search the birth and death parameter (λ)of genes, calculated the probability of each gene family with observed sizes using 10,000 Monte Carlo random samplings, and reported birth and death parameters in gene families with probabilities less than 0.01. For the gene family expansion and contraction analysis in H. comes, we first filtered out gene families without homology in the SWISS-PROT database to reduce the potential false positive expansions or contractions caused by gene prediction. The families that contained sequences that have multiple functional annotations were also removed (Supplementary Tables 4.1 and 4.2).
Phylogenetic analysis
We obtained 4,122 one-to-one orthologous genes from the gene family analysis (Supplementary Information, section 4.1). The protein sequences of one-to-one orthologous genes were aligned using MUSCLE48 with the default parameters. We then filtered the saturated sites and poorly aligned regions using trimAl (ref. 49) with the parameters “-gt 0.8 –st 0.001 –cons 60”. After trimming the saturated sites and poorly aligned regions in the concatenated alignment, 2,128,000 amino acids were used for the phylogenomic analysis. The trimmed protein alignments were used as a guide to align corresponding coding sequences (CDSs). The aligned protein and the fourfold degenerate sites in the CDSs were each concatenated into a super gene using an in-house Perl script.
The phylogenomic tree was reconstructed using RAxML version 8.1.19 (ref. 50) based on concatenated protein sequences. Specifically, we used the PROTGAMMAAUTO parameter to select the optimal amino acid substitution model, specified spotted gar as the outgroup, and evaluated the robustness of the result using 100 bootstraps. To compare the neutral mutation rate of different species, we also generated a phylogeny based on fourfold degenerate sites. The phylogenomic topology was used as input and the “-f e” option in RAxML was used to optimize the branch lengths of the input tree using the alignment of fourfold degenerate sites under the general time reversible (GTR) model as suggested by ModelGenerator version 0.85 (ref. 51). We calculated the pairwise distances to the outgroup (spotted gar) based on the optimized branch length of the neutral tree using the cophenetic.phylo module in the R-package APE52. The Bayesian relaxed-molecular clock (BRMC) method, implemented in the MCMCTree program53, was used to estimate the divergence time between different species. The concatenated CDS of one-to-one orthologous genes and the phylogenomics topology were used as inputs. Two calibration time points based on fossil records, O. latipes–T. nigroviridis (~96.9–150.9 million years ago (Mya)), and D. rerio–G. aculeatus (~149.85–165.2 Mya) (http://www.fossilrecord.net/dateaclade/index.html), were used as constraints in the MCMCTree estimation. Specifically, we used the correlated molecular clock and REV substitution model in our calculation. The MCMC process was run for 5,000,000 steps and sampled every 5,000 steps. MCMCTree suggested that H. comes diverged from the common ancestor of stickleback, Nile tilapia, platyfish, fugu, and medaka approximately 103.8 Mya, which corresponds to the Cretaceous period.
Analysis of OR genes
We downloaded protein sequences of 1,417 OR gene family members from NCBI and mapped them to H. comes genome using Tblastn with “E-value ≤1e-10” and “alignment rate ≥ 0.5”. Solar (in-house software, version 0.9.6) was used to join high-scoring segment pairs (HSPs) between each pair of protein mapping results. We retained alignments with an alignment rate of more than 70% and a mapping identity of more than 40%. Subsequently, the protein sequences were mapped to the genome using GeneWise and extended 280 bp upstream and downstream to define integrated gene models. For phylogenetic analysis, protein sequences were aligned using MUSCLE and a JTT+gamma model was used in a maximum-likelihood analysis using PhyML to construct a phylogenetic tree.
Evidence for loss of tbx4 in H. comes
The synteny analysis of tbx2b-tbx4-brip1 region of H. comes, stickleback, fugu and zebrafish using Vista shows that tbx4 was lost in H. comes (Fig. 3). To exclude the scenario that the absence of tbx4 in the H. comes genome sequence is due to an assembly error, we first validated the micro-synteny region of tbx2b-tbx4-brip1 region in H. comes using a PCR- based genomic walk strategy. Briefly, 28 primer pairs (Supplementary Table 9.1) were designed for overlapping amplicons to ‘walk’ from the end of tbx2b to the start of brip1. Amplicon size and partial end sequencing of these products did not indicate any anomalies in the assembly of the H. comes tbx4 ‘ghost locus’.
In addition, we carried out the following analyses: (1) searched the H. comes genome (TblastN) using Tbx4 protein from zebrafish and Nile tilapia and were unable to find a tbx4 gene; (2) searched the H. comes genome using only the domain sequence of Tbx4 protein but were unable to find a tbx4 gene; (3) searched H. comes and H. erectus transcriptome data for tbx4 (TblastN) using Tbx4 protein from zebrafish and Nile tilapia but were unable to find any matching transcript; (4) searched H. comes and H. erectus transcriptome data with the domain sequence as well and did not find any remnant of a tbx4 gene; and (5) predicted CNEs in the ‘ghost’ tbx4 locus of H. comes using the fugu tbx4 locus as the reference (base) (Supplementary Fig. 9.3). We used the CNEs present in the other fish genome loci (that were absent in H. comes) to search the H. comes genome to rule out the possibility that they may be present elsewhere in the genome. We were unable to find any of these CNEs in the H. comes genome. Finally, we conducted degenerate PCR experiments to ascertain if the tbx4 gene is missing in H. comes. Using a combination of four forward and two reverse primers (Supplementary Table 9.1), we checked for the presence of tbx4 in seven species of Hippocampus (including H. comes and H. erectus), five species of pipefish (four from the genus Syngnathus and one species of Corythoichthys) (all from the family Syngnathidae that lack pelvic fins); ghost pipefish (Solenostomus) and the trumpetfish (Aulostomidae) which are closely related to the Syngnathidae but possess pelvic fins; and five other teleost species that possess pelvic fins (Supplementary Figs 9.1 and 9.2).
Generation of mutant tbx4 zebrafish
We used a CRISPR–Cas9 strategy to generate a tbx4 mutant zebrafish line. Two guide RNAs (gRNAs) were designed targeting zebrafish tbx4 in the 5′ end of the sequence that is upstream of or within the DNA-binding TBOX domain (Supplementary Fig. 9.4). gRNAs were cloned using synthesized oligonucleotides into the pT7gRNA vector as described previously54 (oligonucleotide sequences given in Supplementary Table 9.2). gRNAs were synthesized from this vector after linearization with BamH1-HF (NEB R3136T), transcribed using the MEGAscript T7 Transcription Kit (Thermo Fischer Scientific AM1334) and purified using the mirVana miRNA isolation kit (Thermo Fischer Scientific AM1560). Cas9 mRNA was synthesized from the Cs2+Cas9 vector using the mMessage mMachine Sp6 Transcription Kit (Thermo Fischer Scientific AM1340) and purified using the RNA cleanup protocol from the RNAeasy mini kit (Qiagen 74104).
Zebrafish from a wild caught strain were injected at the one-cell stage with ~50 ng gRNA and ~90 ng Cas9 RNA. These F0 fish were raised to maturity and genotyped using fin clipping, DNA isolation and PCR spanning the target site (genotyping primers given in Supplementary Table 9.2). PCR products were analysed for mutations as described previously54 using T7 endonuclease (NEB M0302L). Mosaic mutant F0 fish were outcrossed to AB wild-type fish and embryos were batch genotyped for transmission of the mutation using PCR and T7 endonuclease. Mutant PCR products were cloned into the pGEM-T vector (Promega, Madison, WI) and sequenced to identify carrier fish transmitting a frameshift mutation. These carrier fish were crossed again to AB wild type and the resulting F1 fish were raised to maturity. The F1 were genotyped using fin clipping, DNA isolation, PCR, T7 endonuclease to identify heterozygous mutant fish followed by cloning and sequencing of the mutant PCR products to validate presence of the frameshift allele. The CRISPR–Cas9 mutation strategy is schematically shown in Extended Data Fig. 5.
In the F0 mutant tbx4 fish we observed pelvic fin loss at low frequency. gRNA#1 gave 3/42 fish with either double- or single-sided pelvic fin loss whereas 1/34 had single-sided pelvic fin loss for gRNA#2 (Extended Data Fig. 5). We observed mutant allele transmission for both gRNA#1 and gRNA#2 but failed to identify a deletion leading to a frameshift mutation for gRNA#2 so no stable line was generated for this CRISPR. For gRNA#1 we identified several frameshift mutants, one of which was further analysed. This mutant has a deletion/replacement mutation in which eight nucleotides are replaced by three nucleotides, leading to an effective 5 bp deletion and the introduction of a frameshift mutation (Extended Data Fig. 5). This mutation introduces a downstream STOP codon leading to a severely truncated protein lacking the DNA binding domain (Supplementary Figs 9.4 and 9.5). The mutant line is maintained on an AB wild-type background.
Loss of CNEs
Using zebrafish as the reference genome, whole-genome alignments of six teleost fishes were generated. The soft-masked genome sequence for zebrafish (Zv9, April 2010) was downloaded from the Ensembl release-75 FTP site. The following soft-masked genome sequences were downloaded from the UCSC Genome Browser: stickleback (gasAcu1, February 2006), fugu (fr3, October 2011), medaka (oryLat2, October 2005), Nile tilapia (oreNil2, February 2012). The H. comes genome sequence (hipCom0) was repeat-masked using WindowMasker (from NCBI BLAST+ package v.2.2.28) with additional parameter “-dust true”. About 32% (158.1/501.6 Mb) of the H. comes genome was masked using this method.
Only chromosome sequences of zebrafish were aligned while unplaced scaffolds were excluded. The reference (zebrafish) genome was split into 21 Mb sequences with 10-kb overlap, while the percomorph fish genomes (H. comes, stickleback, fugu, medaka and Nile tilapia) were split into 10 Mb sequences with no overlap. Pairwise alignments were carried out using Lastz v.1.03.54 (ref. 55) with the following parameters: –strand = both–seed = 12of19–notransition–chain–gapped–gap = 400,30–hspthresh = 3000–gappedthresh = 3000–inner = 2000–masking = 50–ydrop = 9400–scores = HoxD55.q–format = axt. Coordinates of split sequences were restored to genome coordinates using an in-house Perl script. The alignments were reduced to single coverage with respect to the reference genome using UCSC Genome Browser tools ‘axtChain’ and ‘chainNet’. Multiple alignments were generated using Multiz.v11.2/roast.v3 (ref. 56) with the tree topology “(Zv9 (hipCom0 ((fr3 gasAcu1) (oryLat2 oreNil2))))”.
Fourfold degenerate (4D) sites of zebrafish genes (Ensembl release-75) were extracted from the multiple alignments. These 4D sites were used to build a neutral model using PhyloFit in the rphast v.1.5 package57 (general reversible “REV” substitution model). PhastCons was then run in rho-estimation mode on each of the zebrafish chromosomal alignments to obtain a conserved model for each chromosome. These conserved models were averaged into one model using PhyloBoot. Subsequently, conserved elements were predicted in the multiple alignments using PhastCons with the following inputs and parameters: the neutral and conserved models, target coverage of input alignments = 0.3 and average length of conserved sequence = 45 bp. To assess the sensitivity of this approach in identifying functional elements, the PhastCons elements were compared against zebrafish protein-coding genes. Eighty per cent of protein-coding exons (197,508/245,556 exons) were overlapped by a conserved element (minimum coverage 10%), indicating that the identification method was fairly sensitive.
A CNE was considered present in a percomorph genome if it showed coverage of at least 30% with a zebrafish CNE in Multiz alignment. To identify CNEs that could have been missed in the Multiz alignments due to rearrangements in the genomes, or due to partitioning of the CNEs among teleost fish duplicate genes, we searched the zebrafish CNEs against the genome of the percomorph using BLASTN (E < 1 × 10−10; ≥80% identity; ≥30% coverage). Those CNEs that had no significant match in a percomorph genome were considered as missing in that genome. To account for CNEs that might have been missed due to sequencing gaps, we identified gap-free syntenic intervals in zebrafish and the percomorph genomes, and generated a set of CNEs that were missing from these intervals. These CNEs represent a high-confidence set of CNEs missing in the percomorph fishes and thus were used for further analysis. Functional enrichment of genes associated with CNEs was carried out using the GREAT software58 with each CNE assigned to the genes with the nearest transcription start site and within 1 Mb in the zebrafish genome, and significantly enriched functional categories identified based on a hypergeometric test of genomic regions (false discovery rate (FDR) q value < 0.05). We identified the statistically significant gene ontology biological process terms, molecular function terms and zebrafish phenotype descriptions of the genes that are associated with CNEs.
We also predicted CNEs in the Hox clusters of H. comes and other representative teleost fishes using the global alignment program MLAGAN. Orthologous Hox clusters were aligned using MLAGAN with zebrafish as the reference sequence and CNEs were predicted using VISTA.
Functional assay of CNEs
Seven representative zebrafish CNEs that have been lost in H. comes (the largest among the lost CNEs) were assayed for enhancer activity in transgenic zebrafish using GFP as the reporter gene. The CNEs were amplified by PCR using zebrafish genomic DNA as template. The products were cloned into a miniTol2 transposon donor plasmid linked to the mouse cFos (McFos) basal promoter and the coding sequence of GFP. Transposase mRNA was generated by transcribing cDNA in vitro using the mMESSAGE mMACHINE T7 kit (Ambion; Life Technologies). The CNE-containing McFos-miniTol2 construct and transposase mRNA were co-injected into the yolk of zebrafish embryos at the one to two-cell stage. Each CNE construct was injected into 250–350 embryos and the injections were repeated on two days. The embryos were reared at 28 °C, and GFP was observed at 24, 48 and 72 h post-fertilization (hpf). The survival rate of the embryos post-injection was 70–80%. Consistent GFP expression in at least 20% of F0 embryos was considered as specific expression driven by a CNE. Such embryos were reared to maturity and mated with wild type zebrafish to produce F1 lines. The expression of GFP in F1 embryos was observed under a compound microscope fitted for epifluorescence (Axio imager M2; Carl Zeiss, Germany) and photographed using an attached digital microscope camera (Axiocam; Carl Zeiss, Germany). Pigmentation was inhibited by maintaining zebrafish embryos in 0.003% N-phenylthiourea (Sigma-Aldrich, Sweden) from 8 hpf onwards. Consistent GFP expression observed in at least three lines of F1 fishes was considered as the specific expression driven by a CNE.
All animals were cared for in strict accordance with National Institutes of Health (USA) guidelines. The zebrafish gene knockout protocol was approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University. The zebrafish transgenic assay protocol was approved by the Institutional Animal Care and Use Committee of Biological Resource Centre, A*STAR, Singapore.
Data availability statement
The tiger tail seahorse (H. comes) whole-genome sequence has been deposited in the DDBJ/EMBL/GenBank database under accession number LVHJ00000000. RNA-seq reads for H. erectus and H. comes have been deposited in the NCBI Sequence Read Archive under accession numbers SRA392578 and SRA392580, respectively.
References
Leysen, H. et al. Musculoskeletal structure of the feeding system and implications of snout elongation in Hippocampus reidi and Dunckerocampus dactyliophorus . J. Fish Biol. 78, 1799–1823 (2011)
Stölting, K. N. & Wilson, A. B. Male pregnancy in seahorses and pipefish: beyond the mammalian model. BioEssays 29, 884–896 (2007)
Wilson, A. B., Vincent, A., Ahnesjö, I. & Meyer, A. Male pregnancy in seahorses and pipefishes (family Syngnathidae): rapid diversification of paternal brood pouch morphology inferred from a molecular phylogeny. J. Hered. 92, 159–166 (2001)
Teske, P. R., Cherry, M. I. & Matthee, C. A. The evolutionary history of seahorses (Syngnathidae: Hippocampus): molecular data suggest a West Pacific origin and two invasions of the Atlantic Ocean. Mol. Phylogenet. Evol. 30, 273–286 (2004)
Near, T. J. et al. Phylogeny and tempo of diversification in the superradiation of spiny-rayed fishes. Proc. Natl Acad. Sci. USA 110, 12738–12743 (2013)
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989)
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics (Oxford Univ. Press, 2000)
Bailly, X. et al. The loss of the hemoglobin H2S-binding function in annelids from sulfide-free habitats reveals molecular adaptation driven by Darwinian positive selection. Proc. Natl Acad. Sci. USA 100, 5885–5890 (2003)
MacArthur, D. G. et al. Loss of ACTN3 gene function alters mouse muscle metabolism and shows evidence of positive selection in humans. Nature Genet. 39, 1261–1265 (2007)
Kawasaki, K. The SCPP gene family and the complexity of hard tissues in vertebrates. Cells Tissues Organs 194, 108–112 (2011)
Louchart, A. & Viriot, L. From snout to beak: the loss of teeth in birds. Trends Ecol. Evol. 26, 663–673 (2011)
Meredith, R. W., Zhang, G., Gilbert, M. T., Jarvis, E. D. & Springer, M. S. Evidence for a single loss of mineralized teeth in the common avian ancestor. Science 346, 1254390 (2014)
Deméré, T. A., McGowen, M. R., Berta, A. & Gatesy, J. Morphological and molecular evidence for a stepwise evolutionary transition from teeth to baleen in mysticete whales. Syst. Biol. 57, 15–37 (2008)
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320 (2014)
Yamanoue, Y., Setiamarga, D. H. & Matsuura, K. Pelvic fins in teleosts: structure, function and evolution. J. Fish Biol. 77, 1173–1208 (2010)
Kuiter, R. H. Seahorses and their Relatives (Aquatic Photographics, 2009)
Harris, J. E. The role of the fins in the equilibrium of the swimming fish. II. The role of the pelvic fins. J. Exp. Biol. 15, 32–47 (1938)
Gosline, W. A. The evolution of some structural systems with reference to the interrelationships of modern lower teleostean fish groups. Jpn. J. Ichthyol. 27, 1–28 (1980)
Standen, E. M. Pelvic fin locomotor function in fishes: three-dimensional kinematics in rainbow trout (Oncorhynchus mykiss). J. Exp. Biol. 211, 2931–2942 (2008)
Tanaka, M. et al. Developmental genetic basis for the evolution of pelvic fin loss in the pufferfish Takifugu rubripes . Dev. Biol. 281, 227–239 (2005)
Chan, Y. F. et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305 (2010)
Naiche, L. A. & Papaioannou, V. E. Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development 130, 2681–2693 (2003)
Rodriguez-Esteban, C. et al. The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature 398, 814–818 (1999)
Tamura, K., Yonei-Tamura, S. & Izpisúa Belmonte, J. C. Differential expression of Tbx4 and Tbx5 in zebrafish fin buds. Mech. Dev. 87, 181–184 (1999)
Arora, R., Metzger, R. J. & Papaioannou, V. E. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet. 8, e1002866 (2012)
Don, E. K. et al. Genetic basis of hindlimb loss in a naturally occurring vertebrate model. Biol. Open 5, 359–366 (2016)
Kawaguchi, M. et al. Evolution of teleostean hatching enzyme genes and their paralogous genes. Dev. Genes Evol. 216, 769–784 (2006)
Harlin-Cognato, A., Hoffman, E. A. & Jones, A. G. Gene cooption without duplication during the evolution of a male-pregnancy gene in pipefish. Proc. Natl Acad. Sci. USA 103, 19407–19412 (2006)
Whittington, C. M., Griffith, O. W., Qi, W., Thompson, M. B. & Wilson, A. B. Seahorse brood pouch transcriptome reveals common genes associated with vertebrate pregnancy. Mol. Biol. Evol. 32, 3114–3131 (2015)
Kawaguchi, M., Tomita, K., Sano, K. & Kaneko, T. Molecular events in adaptive evolution of the hatching strategy of ovoviviparous fishes. J. Exp. Zool. B Mol. Dev. Evol. 324, 41–50 (2015)
Bejerano, G. et al. Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004)
Lindblad-Toh, K. et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011)
Venkatesh, B. et al. Ancient noncoding elements conserved in the human genome. Science 314, 1892 (2006)
Navratilova, P. et al. Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev. Biol. 327, 526–540 (2009)
Visel, A. et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nature Genet. 40, 158–160 (2008)
Attanasio, C. et al. Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342, 1241006 (2013)
McLean, C. Y. et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471, 216–219 (2011)
Sabherwal, N. et al. Long-range conserved non-coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Hum. Mol. Genet. 16, 210–222 (2007)
Shears, D. J. et al. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri–Weill dyschondrosteosis. Nature Genet. 19, 70–73 (1998)
Indjeian, V. B. et al. Evolving new skeletal traits by cis-regulatory changes in bone morphogenetic proteins. Cell 164, 45–56 (2016)
Lin, Q., Lin, J. & Zhang, D. Breeding and juvenile culture of the lined seahorse, Hippocampus erectus Perry, 1810. Aquaculture 277, 287–292 (2008)
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012)
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnol. 29, 644–652 (2011)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25, 1105–1111 (2009)
Yu, X., Lin, J., Zack, D. J. & Qian, J. Computational analysis of tissue-specific combinatorial gene regulation: predicting interaction between transcription factors in human tissues. Nucleic Acids Res. 34, 4925–4936 (2006)
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002)
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004)
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009)
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006)
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771 (2008)
Paradis, E., Claude, J. & Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004)
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007)
Jao, L. E., Wente, S. R. & Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl Acad. Sci. USA 110, 13904–13909 (2013)
Harris, R. S. Improved Pairwise Alignment of Genomic DNA. PhD thesis, Pennsylvania State Univ. (2007)
Blanchette, M. et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14, 708–715 (2004)
Hubisz, M. J., Pollard, K. S. & Siepel, A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinform. 12, 41–51 (2011)
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnol. 28, 495–501 (2010)
Bian, C. et al. The Asian arowana (Scleropages formosus) genome provides new insights into the evolution of an early lineage of teleosts. Sci. Rep. 6, 24501 (2016)
Kawasaki, K. & Amemiya, C. T. SCPP genes in the coelacanth: tissue mineralization genes shared by sarcopterygians. J. Exp. Zool. B Mol. Dev. Evol. 322, 390–402 (2014)
Venkatesh, B. et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 505, 174–179 (2014)
Acknowledgements
We thank the Laboratory Animal Center of Sun Yat-Sen University for providing experimental facilities and the Agency for Science, Technology and Research (A*STAR) Computational Resource Centre for use of its high-performance computing facilities. This research was supported by the National Science Fund for Excellent Young Scholars (41322038), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020103), the Youth Foundation of National High Technology Research and Development Program (863 Program) (2015AA020909), the Outstanding Youth Foundation in Guangdong Province (S2013050014802), the National Natural Science Foundation of China (41576145), the National Key Basic Research Program of China (2015CB452904), the Special Project on the Integration of Industry, Education and Research of Guangdong Province (no. 2013B090800017) and the Biomedical Research Council of A*STAR, Singapore.
Author information
Authors and Affiliations
Contributions
Q.L., A.M. and B.V. designed the scientific objectives. Q.L., B.V., A.M., Q.S. and Y.Z. oversaw the project. H.Z., G.Q., B.V. and Y.Z. collected samples for sequencing DNA and RNA. M.X., Y.Y., J.M. and Q.G. performed genome sequencing, assembly and annotation. S.F., J.M. and Y.Y. performed phylogenomic analysis and molecular evolutionary rate analysis. S.F. and M.X. characterized repetitive sequences and GC content. S.F., R.F.S., P.X., V.R. and B.V. annotated and analysed Hox clusters. V.R. and B.V. annotated and analysed SCPP genes. A.P.L., V.R., Z.W.L. and B.V. performed CNE analysis and functional assay of zebrafish CNEs. Y.Z., M.X., C.Z. and D.S. assembled and annotated RNA-seq data. H.M.G. and S.F. interpreted RNA-seq results and designed the qRT–PCR experiment. Y.Z., H.Z. and X.W. performed qRT–PCR to validate the expression levels of transcripts. Y.Z., M.X., H.Z. and V.R. analysed the patristacin gene family. Y.Z., Q.L., J.M.W., R.F.S. and A.M. performed tbx4 knockout analysis. Y.Q., J.B., C.B., Y.S. and X.Z. were involved in data analysis. L.H., G.L., W.L., Z.G., K.W. and H.Q. participated in the discussions related to data analysis. Q.L., Y.Z., S.F., H.M.G., A.M. and B.V. wrote the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks C. Amemiya, S. Burgess, K. Worley and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Phylogenetic relationships of ray-finned fishes discussed in this study.
Phylogenetic relationships of ray-finned fishes depicted here are based on the current study and ref. 59. Ray-finned fishes (Actinopterygii) are divided into basal ray-finned fishes (Polypteriformes, Acipenseriformes and Holostei) and teleosts. The latter comprise ~99% of the extant ray-finned fishes. The star represents the teleost-specific genome duplication (TGD) event that occurred in the common ancestor of all teleost fishes. Syngnathids (seahorse and pipefish) display the unique phenomenon of ‘male pregnancy’.
Extended Data Figure 2 Number of gene families in various teleosts and the spotted gar.
a, Venn diagram of shared orthologous gene families in seahorse (H. comes), fugu, zebrafish and stickleback. b, The phylogeny and divergence times of seahorse and other teleost fishes based on analysis of genome-wide one-to-one orthologous protein sequences. The numbers at nodes indicate the number of gene families expanded and contracted at different evolutionary time points.
Extended Data Figure 3 Divergence distribution of transposable elements compared to consensus in the transposable element library.
The divergence rate was calculated between the identified transposable elements (TEs) in the H. comes genome and the consensus sequence in the transposable element library.
Extended Data Figure 4 SCPP genes in H. comes and other jawed vertebrates.
Gene loci for human, coelacanth and zebrafish were adapted from other publications60,61. sparcl1, which is the ancestral gene that gave rise to SCPP genes is shown in grey; P/Q-rich SCPP genes are shown in red; acidic SCPP genes are shown in blue. In seahorse, scpp5 is a pseudogene and is denoted by ψ. Owing to space constraints, the P/Q-rich SCPP genes encoding milk casein and salivary proteins in human have been omitted. Black circles mark the ends of scaffolds.
Extended Data Figure 6 CRISPR–Cas9 mediated knockdown of tbx4 in zebrafish.
a, CRISPR–Cas9 mutagenesis strategy. b, CRISPR–Cas9 sites targeted in zebrafish tbx4 gene. c, Loss of function tbx4 phenotypes in F0 mosaic mutants. Pelvic fin loss was observed with low frequency in F0 mosaic mutant fish. Frequency of animals with either single- or double-sided loss of pelvic fins was 3/42 for gRNA#1 and 1/34 for gRNA#2. d, Identification of zebrafish tbx4 mutant line. Top shows sequencing chromatograms of wild-type (left) and mutant (right) alleles. Bottom shows alignment of tbx4 exon 2 from wild-type and mutant. The region for which the chromatograms are shown is indicated with a box. In the mutant a deletion (indicated in blue in the wild-type sequence)/substitution (indicated in lilac in the mutant sequence) was identified. The deletion/substitution area is indicated with a grey box in the chromatograms.
Extended Data Figure 7 Reporter gene expression pattern driven by zebrafish CNEs that are lost in H. comes.
Lateral and dorsal views of 72 h post-fertilization F1 transgenic zebrafish embryos. The lost CNEs (#6500, #6560, #hoxc-1 and #hoxd-1) were assayed for their reporter gene expression potential in transgenic zebrafish. FB, forebrain; FP, floor plate; H, heart; HB, hindbrain; L, lens; M, melanocytes; MB, midbrain; O, otic vesicle; SC, spinal cord.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data, Supplementary Tables, Supplementary Figures and Supplementary References. (PDF 6916 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, Q., Fan, S., Zhang, Y. et al. The seahorse genome and the evolution of its specialized morphology. Nature 540, 395–399 (2016). https://doi.org/10.1038/nature20595
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature20595
This article is cited by
-
Tbx4 and Tbx5 gene expression associated with appendage development and its relationship with the absence of the pelvic fin in Pampus argenteus
Journal of Oceanology and Limnology (2024)
-
Expanded olfactory system in ray-finned fishes capable of terrestrial exploration
BMC Biology (2023)
-
Expression of secretory calcium-binding phosphoprotein (scpp) genes in medaka during the formation and replacement of pharyngeal teeth
BMC Oral Health (2023)
-
Gene losses, parallel evolution and heightened expression confer adaptations to dedicated cleaning behaviour
BMC Biology (2023)
-
Evolutionary dynamics of genome size and content during the adaptive radiation of Heliconiini butterflies
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
