
Abstract
Zika virus (ZIKV) is an emerging mosquito-transmitted flavivirus that can cause severe disease, including congenital birth defects during pregnancy1. To develop candidate therapeutic agents against ZIKV, we isolated a panel of human monoclonal antibodies from subjects that were previously infected with ZIKV. We show that a subset of antibodies recognize diverse epitopes on the envelope (E) protein and exhibit potent neutralizing activity. One of the most inhibitory antibodies, ZIKV-117, broadly neutralized infection of ZIKV strains corresponding to African and Asian-American lineages. Epitope mapping studies revealed that ZIKV-117 recognized a unique quaternary epitope on the E protein dimer–dimer interface. We evaluated the therapeutic efficacy of ZIKV-117 in pregnant and non-pregnant mice. Monoclonal antibody treatment markedly reduced tissue pathology, placental and fetal infection, and mortality in mice. Thus, neutralizing human antibodies can protect against maternal–fetal transmission, infection and disease, and reveal important determinants for structure-based rational vaccine design efforts.
Similar content being viewed by others

Main
Recent ZIKV epidemics are linked to Guillain–Barré syndrome in adults and microcephaly in fetuses and newborn infants2,3,4,5. Although ZIKV infection can potentially cause severe disease, specific treatments and vaccines for ZIKV are not currently available. We sought to isolate neutralizing human monoclonal antibodies (mAbs) with broad specificity against all ZIKV strains and protective activity in vivo. We tested the serological response of subjects who had previously been infected with ZIKV in diverse geographic locations. Serum from each subject contained antibodies that were shown by ELISA assays to react with ZIKV E protein and to neutralize infection of a contemporary Asian isolate (H/PF/2013) from French Polynesia (Fig. 1a, b). We studied the B cells of subject 1001 in greater detail. Based on the results of replicate assays, the frequency of B cells that secrete antibodies against ZIKV E protein in the peripheral blood was between 0.36% and 0.61% (Fig. 1c, d). We next tested the reactivity of antibodies with domain III (DIII) of the E protein from ZIKV or the related dengue (DENV) and West Nile (WNV) viruses. Only a subset (6%) of the ZIKV-E-reactive antibodies bound to DIII, and most were specific for ZIKV (Fig. 1c). Comparative binding to a wild-type or mutant ZIKV E protein lacking the conserved fusion loop epitope in DII (mutant denoted hereafter as E-FLM) established immunodominance (binding around 70% of mAbs) of the fusion loop.

a, b, Serum samples from humans with a previous ZIKV infection were tested for binding to ZIKV E protein in ELISA (a) (with two technical replicates) and neutralization of ZIKV (b) (at least two independent repeats in triplicate). Subject 1001 had the highest endpoint titre in the binding assay and displayed potent neutralizing activity. Subject 657 was a control without history of exposure to ZIKV. c, Supernatants of Epstein–Barr virus (EBV)-transformed B-cell cultures from subject 1001 were tested for binding to ZIKV E or DIII of ZIKV E or related flavivirus E proteins; the WNV-reactive clone and all but one DENV-reactive B-cell line also reacted with ZIKV E protein. The frequency of antigen-specific cells against each viral protein was determined with a threshold absorbance value at 405 nm (A405 nm) of 1.5 as indicated. d, In four additional separate B-cell transformation experiments, the frequency of B cells reactive with intact ZIKV E or E-FLM was determined.
We obtained 29 cloned hybridomas secreting mAbs that bound to ZIKV E protein from the cells of three donors (mAb ZIKV-195 from subject 1011, mAb ZIKV-204 from subject 973, and the remaining 27 mAbs from subject 1001). All of the mAbs except for one belonged to the IgG1 isotype (two could not be determined), with an equal distribution of κ and λ light chains (Extended Data Table 1); sequence analysis of cDNA of the antibody variable gene regions revealed that each mAb represented an independent clone (Extended Data Table 1). We determined the half-maximal effective concentrations for binding to ZIKV E protein (EC50) and neutralization (IC50) of infection (Fig. 2a, Extended Data Fig. 1); most of the mAbs bound to E protein at low concentrations (EC50 < 100 ng ml−1), whereas only four of the 29 mAbs exhibited strong neutralizing activity (IC50 = 5–420 ng ml−1). We next determined how many antigenic sites on ZIKV E were recognized using quantitative competition binding. We identified four major competition groups (designated A, B, C or D). Group A mAbs had 23 members that were directed against the fusion loop in DII, as determined by differential binding to E and E-FLM (Extended Data Fig. 1), and had only one clone (ZIKV-88) with moderate neutralizing potency. The group B mAb ZIKV-116 neutralized ZIKV infection and bound to E, DIII and E-FLM. Group C mAbs (ZIKV-19 and ZIKV-190) bound to E and E-FLM weakly, but did not potently neutralize infection. The group D mAb ZIKV-195 neutralized with moderate potency and was similar in binding to both E and E-FLM. The most inhibitory group D mAb, ZIKV-117, bound to both E and E-FLM weakly.

a, We tested 29 mAbs in binding, neutralization, and competition binding assays. The EC50 against ZIKV E and the IC50 (by focus reduction neutralization test) against H/PF/2013 strain for neutralizing antibodies (highlighted in blue) are shown. The mAbs are displayed in four groups (A, B, C or D) based on a competition binding assay. The values are the percentage of binding that occurred during competition compared to non-competed binding, which was normalized to 100% and the range of competition is indicated by the box colours. Black filled boxes indicate strongly competing pairs (residual binding <30%), grey filled boxes indicate intermediate competition (residual binding 30–69%), and white filled boxes indicate non-competing pairs (residual binding ≥ 70%). The IC50 against H/PF/2013 strain for neutralizing antibodies is shown with neutralizing clones highlighted in blue. b, A ribbon diagram of three protomers of ZIKV E (DI in red, DII in yellow and DIII in blue) is shown with critical residues highlighted as spheres from epitope mapping experiments for representative antibodies in each of the competition binding groups. The colours of the critical residues correspond to the competition group designation as in a. The mutations in the E-FLM and DIII-LR mutants are indicated by black and silver spheres, respectively. c, Representative mAbs from each competition binding group are listed with the domains and residues critical for binding. FL, fusion loop. d, Two mAbs were tested for neutralization of five strains of ZIKV. The concentrations (ng ml−1) at which 50% or 90% neutralization occurred are listed in e. The neutralization data are pooled from at least three independent experiments performed in triplicate.
We mapped the epitopes of representative mAbs using a shotgun alanine-scanning mutagenesis library6 of ZIKV prM and E protein variants (Fig. 2b, Extended Data Fig. 2). Loss-of-binding analysis confirmed that group A mAbs bound to the fusion loop in DII, whereas the group B mAb bound to DIII. Group B mAb ZIKV-116 bound to an epitope involving residues T309, E393 and K394 along the lateral ridge of DIII (DIII-LR), which was confirmed in an ELISA that showed reduced binding to DIII with mutations A310E and T335K in DIII-LR7. The epitope mapping studies suggest that the group D mAb ZIKV-117 binds specifically to DII across two adjacent dimers at the ‘dimer–dimer’ interface (Fig. 2c). We were unable to isolate virus neutralization escape mutant viruses for ZIKV-117, despite six passages in cell culture under mAb selection pressure.
Because of their potency, we assessed whether group B mAb ZIKV-116 and group D mAb ZIKV-117 could inhibit diverse ZIKV strains encompassing the African and Asian-American lineages. ZIKV-117 neutralized all of the ZIKV strains tested, including two African (MR 766 and Dakar 41519), two Asian (Malaysia P6740 and H/PF/2013), and an American (Brazil Paraiba 2015) strain with IC50 values of 5 to 25 ng ml−1 (Fig. 2d, e). ZIKV-116 inhibited four of the five strains efficiently, but was inactive against MR 766, the original African strain (Fig. 2d, e). Alignment of the sequences of ZIKV H/PF/2013 and MR 766, with respect to residues in DIII-LR7 that ZIKV-116 binds, revealed only one difference (a conservative E393D change). Given these data, we hypothesize that the DIII-LR epitope of ZIKV-116 is displayed differently on MR 766 owing to allosteric effects of changes in other parts of the E protein, which could regulate epitope accessibility8,9.
As recent studies have suggested that cross-reactive ZIKV-specific human mAbs can enhance DENV infection in vivo10, we tested whether these two ZIKV-neutralizing mAbs could bind to DENV-infected cells. ZIKV-117 showed a restricted type-specific binding pattern as it failed to stain cells infected with DENV-1, DENV-2, DENV-3 or DENV-4, or bind to purified WNV E protein (Extended Data Fig. 3 and data not shown). In comparison, ZIKV-116 bound to cells infected with DENV-1, DENV-2 or DENV-4, but did not bind to DENV-2 DIII or WNV DIII in ELISA.
In vivo models of ZIKV pathogenesis and antibody prophylaxis have been reported7,10,11 in mice deficient in type-I interferon signalling. To determine whether ZIKV-117 had therapeutic activity, we treated 4–5-week-old wild-type male C57BL/6 mice at day −1 with anti-Ifnar1 mAbs, and then inoculated animals with 103 focus-forming units (FFU) of a mouse-adapted African strain of ZIKV-Dakar7. Animals were treated with a single dose of ZIKV-117 or non-binding isotype control (human (h)CHK-152)12, on day +1 (100 μg; 6.7 mg kg−1) or day +5 (250 μg; 16.7 mg kg−1). Animals treated with hCHK-152 sustained significant lethality compared to those receiving ZIKV-117 (Fig. 3a), which were protected even when administered only a single dose 5 days after virus inoculation.

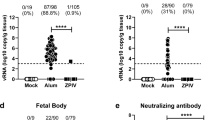
a, We treated 4–5-week-old wild-type male mice with 2 mg of anti-Ifnar1 mAb followed by subcutaneous inoculation with 103 FFU of mouse-adapted ZIKV-Dakar. Mice were treated with a single 100 μg or 250 μg dose of isotype control mAb (hCHK-152) or ZIKV-117 on D+1 or D+5 (n = 10 per group from two independent experiments), respectively. Significance was analysed by the log-rank test (*P < 0.05; **P < 0.01). b, c, Ifnar1−/− female mice were mated with wild-type sires. At E5.5, dams were treated with 250 μg of either hCHK-152 isotype control mAb or ZIKV-117. Bars indicate the median values and reflect data pooled from four independent experiments. Significance for fetal survival and viral RNA was analysed by chi-square (b; ****P < 0.0001) and Mann–Whitney (c; *P < 0.05) tests, respectively. d–f, Wild-type female mice were mated with wild-type sires. At E5.5, dams were treated with anti-Ifnar1 mAb and one of the following: PBS (d, e), 250 μg (d–f) of hCHK-152 isotype control mAb, 250 μg of ZIKV-117 (d–f) or 250 μg of ZIKV-117 LALA (f). At E6.5, dams were inoculated with 103 FFU of ZIKV-Dakar. d–f, Fetuses and placentas (d, f) and maternal brain and serum (e) were collected on E13.5 and viral RNA was measured by qRT–PCR. Bars indicate the median values of samples collected from three biological replicates (d, n = 20–36; e, n = 5–9; f, n = 23–28). Significance was analysed by ANOVA with a Dunn’s multiple comparison test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). g, h, Wild-type female mice were mated with wild-type sires. At E5.5, dams were treated with anti-Ifnar1 mAb. At E6.5, dams were inoculated with 103 FFU of ZIKV-Dakar. At E7.5 (day +1 after infection), dams were treated with PBS, 250 μg of hCHK-152 isotype control mAb, or 250 μg of ZIKV-117. g, h, Fetuses and placentas (g) and maternal brain and serum (h) were collected on E13.5 and viral RNA was measured by qRT–PCR. Bars indicate the median values of samples collected from three biological replicates (g, n = 8–20; h, n = 3–7). Significance was analysed by ANOVA with Dunn’s (g) or Tukey’s (h) multiple comparisons test (*P < 0.05, ***P < 0.001, ****P < 0.0001). Dashed lines indicate the limit of detection of the assay.
We and others have demonstrated placental injury and fetal demise following ZIKV infection of pregnant mice with deficiencies in type-I interferon signalling13,14,15. To assess the protective ability of ZIKV-117 during fetal development, we treated Ifnar1−/− pregnant dams mated to wild-type male mice with a single 250 μg dose of ZIKV-117 or isotype control mAb (hCHK-152) on embryo day 5.5 (E5.5), the day before ZIKV inoculation. Whereas inoculation with ZIKV-Brazil at E6.5 following administration with hCHK-152 resulted in high levels of maternal infection and almost uniform fetal demise by E13.5, treatment with ZIKV-117 improved fetal outcome (Fig. 3b, c).
Because of the extent of demise at E13.5 after ZIKV infection of Ifnar1−/− dams, we could not recover adequate numbers of fetuses to measure viral titres. Accordingly, we switched to a wild-type mouse model with an acquired type-I interferon deficiency using the mouse-adapted African ZIKV-Dakar strain. Wild-type pregnant dams were treated at day −1 (E5.5) with an anti-Ifnar1 mAbs. At the same time, these animals were administered vehicle control (PBS), 250 μg isotype control hCHK-152, or 250 μg ZIKV-117. One day later, dams were inoculated subcutaneously with 103 FFU of ZIKV-Dakar. Fetuses from dams treated with anti-Ifnar1 mAbs and given PBS or hCHK-152 showed high levels (for example, around 105 to 107 FFU equivalents per gram) of viral RNA in the placenta and fetal brain (Fig. 3d). In comparison, mice treated with anti-Ifnar1 and ZIKV-117 had reduced virus levels in the placenta and fetal brain (for example, around 10° to 103 FFU equivalents per gram). This phenotype was associated with transport of human ZIKV E-specific IgG across the maternal–fetal placental barrier (816 ± 53 ng ml−1 for the placenta and 1,675 ± 203 ng ml−1 for the fetal head; Extended Data Fig. 4). As levels of neonatal Fc receptor in the mouse placenta are lower than other mammalian species16, reduced levels of transport of maternal or exogenous IgG into the fetus is expected17. Although this factor could underestimate the therapeutic effect of exogenous anti-ZIKV IgG or maternal antibodies, we nonetheless achieved levels in the placenta and fetal head that were orders of magnitude above the IC50 neutralization value for ZIKV-117. Dams treated with ZIKV-117 also had substantially lower levels of viral RNA in the maternal brain and serum (Fig. 3e).
Antibody-dependent enhancement of flavivirus infection occurs when type-specific or cross-reactive antibodies fail to reach a stoichiometric threshold for neutralization and instead facilitate infection of FcγR-expressing myeloid cells18. Because antibodies can promote antibody-dependent enhancement of ZIKV in cell culture19,20, we evaluated the protective efficacy of a recombinant form of ZIKV-117 IgG containing a leucine (L) to alanine (A) substitution at positions 234 and 235 (LALA)21, which lacked efficient binding to FcγR, retained interactions with FcRn22, and neutralized ZIKV in vitro equivalently compared to the parent mAb (Extended Data Fig. 5). The LALA variant of ZIKV-117 showed similar protective activity against infection of the placenta and fetus relative to the parent mAb (Fig. 3f). As the protection conferred by ZIKV-117 in the pregnancy model is probably due to neutralization and not Fc effector functions, LALA variants could be used without a risk of antibody-dependent enhancement.
We next assessed the post-exposure efficacy of ZIKV-117 during pregnancy. Mice treated with anti-Ifnar1 mAbs at E5.5 were inoculated with 103 FFU of ZIKV-Dakar at E6.5 and then administered a single dose of PBS, 250 μg of hCHK-152, or 250 μg of ZIKV-117 at E7.5. Compared to PBS or isotype control mAb treatment, administration of ZIKV-117 markedly reduced the viral burden in the dams, the placenta and fetus when measured at E13.5 (Fig. 3g, h).
The reduction in viral load mediated by ZIKV-117 was associated with decreased damage of the placenta (as judged by labyrinth layer and overall placenta area), less trophoblast cell death, and increased body size of the fetus (Fig. 4a–c) compared to fetuses of PBS- or hCHK-152-treated dams. ZIKV-117 protected against ZIKV-induced placental insufficiency, as the placental area and fetal size from infected dams treated with anti-ZIKV mAbs were similar to that of uninfected placentas14. In situ hybridization revealed an almost complete absence of viral RNA in the junctional zone and decidua of the placenta in animals treated with ZIKV-117 compared to staining observed in PBS- or hCHK-152-treated controls (Fig. 4d, Extended Data Fig. 6). We also observed vascular damage associated with ZIKV infection of the placenta14, characterized as diminished vimentin staining of fetal endothelial cells, which was rescued by ZIKV-117 to levels seen in uninfected placentas (Fig. 4e). The histopathological data suggests that ZIKV-117 treatment can reduce the ability of ZIKV to cross the fetal endothelial cell barrier, and thereby prevent vertical transmission and improve fetal outcome.

a, Cartoon depicting murine placental structures and zones. b–e, Pregnant dams were treated with PBS, hCHK-152, or ZIKV-117 as described in Fig. 3d–f before infection with ZIKV-Dakar or mock-infected. b, Haematoxylin and eosin staining of placenta at E13.5. Placental labyrinth zone is marked with a solid line. Low power (scale bar, 1 mm) and high power (scale bar, 50 μm) images are presented in sequence. Black arrows indicate apoptotic trophoblasts in areas corresponding to regions of ZIKV infectivity (see panel d, below). c, Measurements of thickness and indicated areas of placenta and fetus body size. Each symbol represents data from an individual placenta or fetus. Significance was analysed by ANOVA with a Dunn’s multiple comparison test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, P > 0.05, NS, not significant). d, In situ hybridization. Low (scale bar, 500 μm) and high (scale bar, 50 μm) power images are presented in sequence. Black arrows indicate cells positive for ZIKV RNA in the junctional zone of the placenta. The images in panels are representative of several placentas from independent dams. e, Low (scale bar, 50 μm) and high (scale bar, 10 μm) power magnified images of immunofluorescence staining of placentas for vimentin (in green, which marks fetal capillary endothelium) from ZIKV-infected dams treated with PBS or ZIKV-117 or from uninfected pregnant animals. Nuclei are counter-stained blue with DAPI.
Our most potent neutralizing antibodies exhibited a breadth of inhibitory activity against strains from Africa, Asia, and the Americas. Even a single ZIKV-117 dose given 5 days after infection protected mice against lethal infection, a timeline similar to the most protective antibodies against other flaviviruses23. Prophylaxis or post-exposure therapy of pregnant mice with ZIKV-117 reduced infection in mothers, and in placental and fetal tissues. As the extent to which these observations in mice translate to humans remains unclear, protection studies in non-human primates, which share a placental architecture similar to humans, seem warranted. If the results were consistent, ZIKV-117 or human antibodies with similar profiles10,19 could be developed as a treatment measure during pregnancy for at-risk humans. By defining key epitopes on the E protein associated with antibody-mediated protection, our studies also inform vaccine efforts to design new immunogens that elicit highly protective antibody responses against ZIKV.
Methods
Research subjects
We studied eight subjects in the United States with previous or recent ZIKV infection (Extended Data Table 2). The studies were approved by the Institutional Review Board of Vanderbilt University Medical Center; samples were obtained after informed consent was obtained by the Vanderbilt Clinical Trials Center. Two subjects (972 and 973) were infected with an African lineage strain in 2008 (one subject while working in Senegal, the second acquired the infection by sexual transmission from the first, as previously reported24). The other six subjects were infected during the current outbreak of an Asian lineage strain, following exposure in Brazil, Mexico or Haiti.
Generation and quantification of human B-cell lines secreting ZIKV E protein specific antibodies
Peripheral blood mononuclear cells (PBMCs) from heparinized blood were isolated with Ficoll-Histopaque by density gradient centrifugation. The cells were used immediately or cryopreserved in the vapour phase of liquid nitrogen until use. Ten million PBMCs were cultured in 384-well plates (Nunc) using culture medium (ClonaCell-HY Medium A, StemCell Technologies) supplemented with 8 μg ml−1 of the TLR agonist CpG (phosphorothioate-modified oligodeoxynucleotide ZOEZOEZZZZZOEEZOEZZZT, Invitrogen), 3 μg ml−1 of Chk2 inhibitor (Sigma), 1 μg ml−1 of cyclosporine A (Sigma), and clarified supernatants from cultures of B95.8 cells (ATCC) containing Epstein–Barr virus. After 7 days, cells from each 384-well culture plate were expanded into four 96-well culture plates (Falcon) using ClonaCell-HY Medium A containing 8 μg ml−1 of CpG, 3 μg ml−1 of Chk2 inhibitor, and 107 irradiated heterologous human PBMCs (Nashville Red Cross) and cultured for an additional 4 days. Supernatants were screened in ELISA (described below) for reactivity with various ZIKV E proteins, which are described below. The minimal frequency of ZIKV E-reactive B cells was estimated based on the number of wells with E protein-reactive supernatants compared with the total number of lymphoblastoid cell line colonies in the transformation plates (calculation: E-reactive B-cell frequency = (number of wells with E-reactive supernatants) divided by (number of LCL colonies in the plate) × 100).
Protein expression and purification
The ectodomains of ZIKV E (H/PF/2013; GenBank Accession KJ776791) and the fusion-loop mutant E-FLM (containing four mutations: T76A, Q77G, W101R, L107R) were expressed transiently in Expi293F cells and purified as described previously7. ZIKV DIII (residues 299–407 of strain H/PF/2013), WNV DIII (residues 296–405 of strain New York 1999) and DENV-2 DIII (residues 299-410 of strain 16681) were expressed in BL21 (DE3) as inclusion bodies and refolded in vitro25. Briefly, inclusion bodies were denatured and refolded by gradual dilution into a refolding buffer (400 mM l-arginine, 100 mM Tris (pH 8.3), 2 mM EDTA, 5 and 0.5 mM reduced and oxidized glutathione) at 4 °C. Refolded proteins were purified by size-exclusion chromatography using a Superdex 75, 16/60 (GE Healthcare).
Generation of human hybridomas
Cells from wells with transformed B cells containing supernatants that exhibited reactivity to ZIKV E protein were fused with HMMA2.5 myeloma cells (gift from L. Cavacini) using an established electrofusion technique26. After fusion, hybridomas were suspended in a selection medium containing 100 μM hypoxanthine, 0.4 μM aminopterin, 16 μM thymidine (HAT Media Supplement, Sigma), and 7 μg ml−1 ouabain (Sigma) and cultured in 384-well plates for 18 days before screening hybridomas for antibody production by ELISA. After fusion with HMMA2.5 myeloma cells, hybridomas producing ZIKV E-specific antibodies were cloned biologically by single-cell fluorescence-activated cell sorting. Hybridomas were expanded in post-fusion medium (ClonaCell-HY Medium E, STEMCELL Technologies) until 50% confluent in 75-cm2 flasks (Corning).
For antibody production, cells from one 75-cm2 flask were collected with a cell scraper and expanded to four 225-cm2 flasks (Corning) in serum-free medium (Hybridoma-SFM, Life Technologies). After 21 days, supernatants were clarified by centrifugation and filtered using 0.45-μm pore size filter devices. HiTrap Protein G or HiTrap MabSelectSure columns (GE Healthcare Life Sciences) were used to purify antibodies from filtered supernatants.
Sequence analysis of antibody variable region genes
Total cellular RNA was extracted from pelleted cells from hybridoma clones, and an RT–PCR reaction was performed using mixtures of primers designed to amplify all heavy-chain or light-chain antibody variable regions27. The generated PCR products were purified using AMPure XP magnetic beads (Beckman Coulter) and sequenced directly using an ABI3700 automated DNA sequencer. The variable region sequences of the heavy and light chains were analysed using the IMGT/V-Quest program28,29.
ELISA and EC50 binding analysis
Wells of microtitre plates were coated with purified, recombinant ectodomain of ZIKV E, DIII, DIII-LR mutants (DIII containing A310E and T335K mutations) or DIII of related flaviviruses DENV-2 or WNV and incubated at 4 °C overnight. In ELISA studies with purified mAbs, we used recombinant ZIKV E protein ectodomain with His6 tag produced in Sf9 insect cells (Meridian Life Sciences R01635). Plates were blocked with 5% skimmed milk in PBS-T for 1 h. B-cell culture supernatants or purified antibodies were added to the wells and incubated for 1 h at ambient temperature. The bound antibodies were detected using goat anti-human IgG (γ-specific) conjugated with alkaline phosphatase (Southern Biotech) and pNPP disodium salt hexahydrate substrate (Sigma). In ELISAs that assessed binding of mAbs to DIII and DIII LR mutants, we used previously described murine mAbs ZV-2 and ZV-54 (ref. 7) as controls. A goat anti-mouse IgG conjugated with alkaline phosphatase (Southern Biotech) was used for detection of these antibodies. Colour development was monitored at 405 nm in a spectrophotometer (Biotek). For determining EC50, microtitre plates were coated with ZIKV E or E-FLM that eliminated interaction of fusion-loop specific antibodies. Purified antibodies were diluted serially and applied to the plates. Bound antibodies were detected as above. A nonlinear regression analysis was performed on the resulting curves using Prism (GraphPad) to calculate EC50 values.
ELISA for detection of human antibodies in murine tissues
Fetal head and placental tissues were collected at E13.5 from groups treated with ZIKV-117 or PBS (as a negative control), homogenized in PBS (250 μl) and stored at −20 °C. ELISA plates were coated with ZIKV E protein, and thawed, clarified tissue homogenates were applied undiluted in triplicate. Bound antibodies were detected using goat anti-human IgG (Fc-specific) antibody conjugated with alkaline phosphatase. The quantity of antibody was determined by comparison with a standard curve constructed using purified ZIKV-117 in a dilution series.
Biolayer interferometry competition binding assay
His6-tagged ZIKV E protein was immobilized on anti-His coated biosensor tips (Pall) for 2 min on an Octet Red biosensor instrument. After measuring the baseline signal in kinetics buffer (PBS, 0.01% BSA, and 0.002% Tween 20) for 1 min, biosensor tips were immersed into the wells containing first antibody at a concentration of 10 μg ml−1 for 7 min. Biosensors then were immersed into wells containing a second mAb at a concentration of 10 μg ml−1 for 7 min. The signal obtained for binding of the second antibody in the presence of the first antibody was expressed as a percentage of the uncompeted binding of the second antibody that was derived independently. The antibodies were considered competing if the presence of first antibody reduced the signal of the second antibody to less than 30% of its maximal binding and non-competing if the signal was greater than 70%. A level of 30–70% was considered intermediate competition.
Shotgun mutagenesis epitope mapping
Epitope mapping was performed by shotgun mutagenesis essentially as described previously6. A ZIKV prM/E protein expression construct (based on ZIKV strain SPH2015) was subjected to high-throughput alanine scanning mutagenesis to generate a comprehensive mutation library. Each residue within prM/E was changed to alanine, with alanine codons mutated to serine. In total, 672 ZIKV prM/E mutants were generated (100% coverage), sequence confirmed, and arrayed into 384-well plates. Each ZIKV prM/E mutant was transfected into HEK-293T cells and allowed to express for 22 h. Cells were fixed in 4% (v/v) paraformaldehyde (Electron Microscopy Sciences), and permeabilized with 0.1% (w/v) saponin (Sigma-Aldrich) in PBS plus calcium and magnesium (PBS++). Cells were incubated with purified mAbs diluted in PBS++, 10% normal goat serum (Sigma), and 0.1% saponin. Primary antibody screening concentrations were determined using an independent immunofluorescence titration curve against wild-type ZIKV prM/E to ensure that signals were within the linear range of detection. Antibodies were detected using 3.75 μg ml−1 of AlexaFluor488-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) in 10% NGS/0.1% saponin. Cells were washed three times with PBS++/0.1% saponin followed by two washes in PBS. Mean cellular fluorescence was detected using a high-throughput flow cytometer (HTFC, Intellicyt). Antibody reactivity against each mutant prM/E clone was calculated relative to wild-type prM/E protein reactivity by subtracting the signal from mock-transfected controls and normalizing to the signal from wild-type prM/E-transfected controls. Mutations within clones were identified as critical to the mAb epitope if they did not support reactivity of the test MAb, but supported reactivity of other ZIKV antibodies. This counter-screen strategy facilitates the exclusion of prM/E mutants that are locally misfolded or have an expression defect.
Vertebrate animal studies ethics statement
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance number A3381-01). Inoculations were performed under anaesthesia induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering. No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Viruses and cells
ZIKV strain H/PF/2013 (French Polynesia, 2013) was obtained from X. de Lamballerie (Aix Marseille Université). ZIKV Brazil Paraiba 2015 was provided by S. Whitehead (Bethesda) and originally obtained from P. F. C. Vasconcelos (Instituto Evandro Cargas). ZIKV MR 766 (Uganda, 1947), Malaysia P6740 (1966), and Dakar 41519 (Senegal, 1982) were provided by the World Reference Center or Emerging Viruses and Arboviruses (R. Tesh, University of Texas Medical Branch). Nicaraguan DENV strains (DENV-1 1254-4, DENV-2 172-08, DENV-3 N2845-09, and DENV-4 N703-99) were provided generously by E. Harris (University of California, Berkeley). Virus stocks were propagated in C6/36 Aedes albopictus cells (DENV) or Vero cells (ZIKV). ZIKV Dakar 41519 (ZIKV-Dakar) was passaged twice in vivo in Rag1−/− mice (M. Gorman and M. Diamond, unpublished data) to create a mouse-adapted strain. Virus stocks were titrated by focus-forming assay (FFA) on Vero cells. All cell lines were checked regularly for mycoplasma contamination and were negative. Cell lines were authenticated at acquisition with short tandem repeat method profiling; Vero cells, though commonly misidentified in the field, were used as they are the standard cell line for flavivirus titration.
Neutralization assays
Serial dilutions of mAbs were incubated with 102 FFU of different ZIKV strains (MR 766, Dakar 41519, Malaysia P6740, H/PF/2013, or Brazil Paraiba 2015) for 1 h at 37 °C. The mAb–virus complexes were added to Vero cell monolayers in 96-well plates for 90 min at 37 °C. Subsequently, cells were overlaid with 1% (w/v) methylcellulose in MEM supplemented with 4% heat-inactivated FBS. Plates were fixed 40 h later with 1% PFA in PBS for 1 h at room temperature. The plates were incubated sequentially with 500 ng ml−1 mouse anti-ZIKV (ZV-16, E.F. and M.S.D., unpublished data) and horseradish-peroxidase-conjugated goat anti-mouse IgG in PBS supplemented with 0.1% (w/v) saponin (Sigma) and 0.1% BSA. ZIKV-infected cell foci were visualized using TrueBlue peroxidase substrate (KPL) and quantitated on an ImmunoSpot 5.0.37 macroanalyzer (Cellular Technologies).
mAb binding to ZIKV- or DENV-infected cells
C6/36 Aedes albopictus cells were inoculated with a MOI 0.01 of ZIKV (H/PF/2013) or different DENV serotypes (Nicaraguan strains DENV-1 1254-4, DENV-2 172-08, DENV-3 N2845-09, DENV-4 N703-99). At 120 h post infection, cells were fixed with 4% PFA diluted in PBS for 20 min at room temperature and permeabilized with HBSS supplemented with 10 mM HEPES, 0.1% saponin and 0.025% NaN3 for 10 min at room temperature. 50,000 cells were transferred to U-bottom plates and incubated for 30 min at 4 °C with 5 μg ml−1 of anti-ZIKV human mAbs or negative (hCHK-152)12, or positive (hE60)30 isotype controls. After washing, cells were incubated with Alexa-Fluor-647-conjugated goat anti-human IgG (Invitrogen) at 1:500, fixed in 1% PFA in PBS, processed on MACSQuant Analyzed (Miltenyi Biotec), and analysed using FlowJo software (Tree Star).
Recombinant antibody expression and purification
Total RNA was extracted from hybridoma cells and genes encoding the VH and VL domains were amplified in RT–PCR using IgExp primers31. The PCR products were directly cloned into antibody expression vectors containing the constant domains of wild-type γ1 chain, LALA mutant (leucine (L) to alanine (A) substitution at positions 234 and 235) γ1 chain for the VH domains, and wild-type κ chain for the VL domain in an isothermal amplification reaction (Gibson reaction)32. Plasmids encoding the heavy and light chain were transfected into 293F cells and full-length recombinant IgG was secreted into transfected cell supernatants. Supernatants were collected and IgG purified using Protein G chromatography and eluted into PBS. The functional abrogation of the binding of the LALA variant IgG was confirmed in an ELISA binding assay with recombinant human FcγRI. The binding of wild-type ZIKV-117 or LALA antibody to FcγRI was evaluated, in comparison with the binding pattern of control antibodies (human mAb CKV063 (ref. 33) LALA mutated IgG).
Adult mouse lethal protection experiments
C57BL/6 male mice (4–5-week-old, Jackson Laboratories) were inoculated with 103 FFU of mouse-adapted ZIKV-Dakar by subcutaneous route in the footpad. One-day before infection, mice were treated with 2 mg anti-Ifnar1 mAb (MAR1-5A3, Leinco Technologies) by intraperitoneal injection. ZIKV-specific human mAb (ZIKV-117) or an isotype control (hCHK-152) was administered as a single dose at day +1 (100 μg) or day +5 (250 μg) after infection through an intraperitoneal route. Animals were monitored for 21 days.
Pregnant mouse protection experiments
Wild-type C57BL/6 mice were bred in a specific pathogen-free facility at Washington University School of Medicine. (1) Ifnar1−/− dams, prophylaxis studies: Ifnar1−/− female and wild-type male mice were mated; at E5.5, dams were treated with a single 250 μg dose of ZIKV mAb or isotype control by intraperitoneal injection. At E6.5, mice were inoculated with 103 FFU of ZIKV Brazil Paraiba 2015 by subcutaneous injection in the footpad. (2) Wild-type dams, prophylaxis studies: wild-type female and male mice were mated; at embryonic days E5.5, dams were treated with a single 250 μg dose of ZIKV mAb or isotype control by intraperitoneal injection as well as a 1 mg injection of anti-Ifnar1 (MAR1-5A3). At E6.5, mice were inoculated with 103 FFU of mouse-adapted ZIKV-Dakar by subcutaneous injection in the footpad. At E7.5, dams received a second 1 mg dose of anti-Ifnar1 through an intraperitoneal route. (3) Wild-type dams, therapy studies: wild-type female and male mice were mated; at embryonic days E5.5, dams were treated with a 1 mg injection of anti-Ifnar1 (MAR1-5A3). At E6.5, mice were inoculated with mouse-adapted 103 FFU of ZIKV-Dakar by subcutaneous injection in the footpad. At E7.5, dams received a second 1 mg dose of anti-Ifnar1 as well as a single 250 μg dose of ZIKV mAb or isotype control through an intraperitoneal route. All animals were euthanized at E13.5, and placentas, fetuses and maternal tissues were collected. Fetus size was measured as the crown-rump length × occipitofrontal diameter of the head.
Measurement of viral burden
ZIKV-infected tissues were weighed and homogenized with stainless steel beads in a Bullet Blender instrument (Next Advance) in 200 μl of PBS. Samples were clarified by centrifugation (2,000g for 10 min). All homogenized tissues from infected animals were stored at −20 °C. Tissue samples and serum from ZIKV-infected mice were extracted with RNeasy 96 Kit (tissues) or Viral RNA Mini Kit (serum) (Qiagen). ZIKV RNA levels were determined by TaqMan one-step quantitative reverse transcriptase PCR (qRT–PCR) on an ABI7500 Fast Instrument using published primers and conditions34. Viral burden was expressed on a log10 scale as viral RNA equivalents per g or ml after comparison with a standard curve produced using serial tenfold dilutions of ZIKV RNA.
Viral RNA in situ hybridization
RNA in situ hybridization was performed with RNAscope 2.5 (Advanced Cell Diagnostics) according to the manufacturer’s instructions. PFA-fixed paraffin embedded placental sections were deparaffinized by incubation for 60 min at 60 °C. Endogenous peroxidases were quenched with H2O2 for 10 min at room temperature. Slides were boiled for 15 min in RNAscope Target Retrieval Reagents and incubated for 30 min in RNAscope Protease Plus before probe hybridization. The probe targeting ZIKV RNA was designed and synthesized by Advanced Cell Diagnostics (catalogue number 467771). Negative (targeting bacterial gene dapB) control probes were also obtained from Advanced Cell Diagnostics (catalogue number 310043). Tissues were counterstained with Gill’s haematoxylin and visualized with standard bright-field microscopy.
Histology and immunohistochemistry
Collected placentas were fixed in 10% neutral buffered formalin at room temperature and embedded in paraffin. At least three placentas from different litters with the indicated treatments were sectioned and stained with haematoxylin and eosin to assess morphology. Surface area and thickness of placenta and different layers were measured using Image J software. For immunofluorescence staining on mouse placentas, deparaffinized tissues were blocked in blocking buffer (1% BSA, 0.3% Triton, PBS) for 2 h and incubated with anti-vimentin antibody (1:500, rabbit, Abcam ab92547). Secondary antibody conjugated with Alexa 488 (1:500 in PBS) was applied for 1 h at room temperature. Samples were counterstained with DAPI (4′6′-diamidino-2-phenilindole, 1:1,000 dilution).
Statistical analysis
All virological data were analysed with GraphPad Prism software. Kaplan–Meier survival curves were analysed by the log rank test, and viraemia was compared using an ANOVA with a multiple comparisons test. P < 0.05 indicated statistically significant differences.
Data availability
All relevant data are included with the manuscript; source data for each of the main text figures is provided.
Accession codes
References
Coyne, C. B. & Lazear, H. M. Zika virus—reigniting the TORCH. Nat. Rev. Microbiol. 14, 707–715 (2016)
Oehler, E. et al. Zika virus infection complicated by Guillain-Barré syndrome–case report, French Polynesia, December 2013. Eur. Commun. Dis. Bull. 19, 7–9 (2014)
Musso, D., Nilles, E. J. & Cao-Lormeau, V. M. Rapid spread of emerging Zika virus in the Pacific area. Clin. Microbiol. Infect. 20, O595–O596 (2014)
Araujo, A. Q. C., Silva, M. T. T. & Araujo, A. P. Q. C. Zika virus-associated neurological disorders: a review. Brain 139, 2122–2130 (2016)
Gatherer, D. & Kohl, A. Zika virus: a previously slow pandemic spreads rapidly through the Americas. J. Gen. Virol. 97, 269–273 (2016)
Davidson, E. & Doranz, B. J. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143, 13–20 (2014)
Zhao, H. et al. Structural basis of Zika virus-specific antibody protection. Cell 166, 1016–1027 (2016)
Dowd, K. A., DeMaso, C. R. & Pierson, T. C. Genotypic differences in dengue virus neutralization are explained by a single amino acid mutation that modulates virus breathing. MBio 6, e01559–159 (2015)
Dowd, K. A., Mukherjee, S., Kuhn, R. J. & Pierson, T. C. Combined effects of the structural heterogeneity and dynamics of flaviviruses on antibody recognition. J. Virol. 88, 11726–11737 (2014)
Stettler, K. et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826 (2016)
Swanstrom, J. A. et al. Dengue virus envelope dimer epitope monoclonal antibodies isolated from dengue patients are protective against Zika virus. MBio 7, e01123–16 (2016)
Pal, P. et al. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 9, e1003312 (2013)
Mysorekar, I. U. & Diamond, M. S. Modeling Zika virus infection in pregnancy. N. Engl. J. Med. 375, 481–484 (2016)
Miner, J. J. et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165, 1081–1091 (2016)
Yockey, L. J. et al. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166, 1247–1256.e4 (2016)
Kim, J. et al. FcRn in the yolk sac endoderm of mouse is required for IgG transport to fetus. J. Immunol. 182, 2583–2589 (2009)
Pentšuk, N. & van der Laan, J. W. An interspecies comparison of placental antibody transfer: new insights into developmental toxicity testing of monoclonal antibodies. Birth Defects Res. B Dev. Reprod. Toxicol. 86, 328–344 (2009)
Pierson, T. C. et al. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1, 135–145 (2007)
Dejnirattisai, W. et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with Zika virus. Nat. Immunol. 17, 1102–1108 (2016)
Charles, A. S. & Christofferson, R. C. Utility of a dengue-derived monoclonal antibody to enhance Zika infection in vitro . PLoS Curr. 8, 1–31 (2016)
Hessell, A. J. et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104 (2007)
Oliphant, T. et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 11, 522–530 (2005)
Foy, B. D. et al. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 17, 880–882 (2011)
Nelson, C. A., Lee, C. A. & Fremont, D. H. Oxidative refolding from inclusion bodies. Methods Mol. Biol. 1140, 145–157 (2014)
Yu, X., McGraw, P. A., House, F. S. & Crowe, J. E., Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J. Immunol. Methods 336, 142–151 (2008)
Thornburg, N. J. et al. Human antibodies that neutralize respiratory droplet transmissible H5N1 influenza viruses. J. Clin. Invest. 123, 4405–4409 (2013)
Brochet, X., Lefranc, M.-P. & Giudicelli, V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 36, W503–W508 (2008)
Giudicelli, V. & Lefranc, M. P. IMGT/junctionanalysis: IMGT standardized analysis of the V-J and V-D-J junctions of the rearranged immunoglobulins (Ig) and T cell receptors (TR). Cold Spring Harb. Protoc. 2011, 716–725 (2011)
Williams, K. L. et al. Therapeutic efficacy of antibodies lacking FcγR against lethal dengue virus infection is due to neutralizing potency and blocking of enhancing Antibodies. PLoS Pathog. 9, e1003157 (2013)
Thornburg, N. J. et al. H7N9 influenza virus neutralizing antibodies that possess few somatic mutations. J. Clin. Invest. 126, 1482–1494 (2016)
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009)
Fong, R. H. et al. Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. J. Virol. 88, 14364–14379 (2014)
Lanciotti, R. S. et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 14, 1232–1239 (2008)
Acknowledgements
We thank N. Murphy, J. Govero, M. Gorman, J. Miner, R. Fong and S. Reddy for technical help and advice on experiments. This work was supported by US N.I.H. grants R01 AI073755 (to M.S.D., D.H.F and J.E.C.), R01 AI104972 (to M.S.D.), US N.I.H. contracts HHSN272201400024C (to J.E.C.), HHSN272201400058C (to B.J.D.), HHSN272201400018C (to D.H.F, M.S.D, and J.E.C) and HHSN272201200026C (CSGID; to D.H.F), and by a Preventing Prematurity Initiative grant from the Burroughs Wellcome Fund and an Investigator award from the March of Dimes (to I.U.M.). E.F. was supported by an N.I.H. Pre-doctoral training grant award (T32 AI007163).
Author information
Authors and Affiliations
Contributions
G.S., E.F., I.U.M., B.J.D., M.S.D. and J.E.C. planned the studies. G.S., E.F., N.K., J.M.F., R.G.B., B.C., A.L.B., T.B. and E.D. conducted experiments. H.Z., C.A.N. and D.H.F. provided protein reagents. G.S., E.F., M.S.D., B.C., B.J.D., I.U.M. and J.E.C. interpreted the studies. G.S., E.F., M.S.D. and J.E.C. wrote the first draft of the paper. D.H.F., B.J.D., M.S.D. and J.E.C. obtained funding. All authors reviewed, edited and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
A.L.B., T.B., E.D. and B.J.D. are employees of Integral Molecular. B.J.D. is a shareholder of Integral Molecular. M.S.D. is a consultant for Inbios, Visterra, and Takeda Pharmaceuticals, and on the Scientific Advisory Boards of Moderna and OraGene. J.E.C. is a consultant for Sanofi and Ridgeback Biotherapeutics, is on the Scientific Advisory Boards of PaxVax, CompuVax, GigaGen, Meissa Vaccines, and is a recipient of research grants from Moderna and Sanofi.
Extended data figures and tables
Extended Data Figure 1 Binding of human mAbs to Zika E protein, E DIII or E-FLM.
mAbs are organized by competition binding groups A to D. Purified mAbs were tested for binding to different antigens as indicated in ELISA as described in Methods. Non-linear regression analysis of the data was performed, and the data plotted are the mean and s.d.
Extended Data Figure 2 High resolution epitope mapping of ZIKV mAbs.
a, An alanine scanning mutation library for ZIKV envelope protein was constructed, in which each amino acid of prM/E was mutated individually to alanine (and alanine to serine) and expression constructs arrayed into 384-well plates, one mutation per well. Each clone in the ZIKV prM/E mutation library, expressed in HEK-293T cells, was tested for immunoreactivity with five mAbs from competition groups A–D, measured using an Intellicyt high-throughput flow cytometer. Shown here for each of the five mAbs is the reactivity with the ZIKV E protein mutants that identified the epitope residues for these mAbs. mAb reactivity for each alanine mutant are expressed as percent of the reactivity of mAb with wild-type ZIKV prM/E. Clones with reactivity <30% relative to wild-type ZIKV prM/E were identified as critical for mAb binding. Bars represent the mean and range of at least two replicate data points. Binding of group B mAbs, ZIKV-116 to wild-type ZIKV E DIII (b) or DIII LR mutant (c) was compared with mouse mAbs ZV-2 and ZV-54. Binding of ZIKV-116 was decreased by mutations in DIII-LR. Data plotted are mean ± s.d.
Extended Data Figure 3 Binding of human mAbs to permeabilized DENV-infected C6/36 cells.
C6/36 cells were infected with DENV-1, DENV-2, DENV-3, DENV-4 or mock infected. Cells were stained with the indicated anti-ZIKV mAbs, an isotype negative control (hCHK-152), or a positive control (a cross-reactive antibody to DENV; chimeric human E60 (chE60)) and processed by flow cytometry. The data are representative of two independent experiments. The numbers in the box indicate the fraction of cells that stained positively.
Extended Data Figure 4 Detection of human IgG in placenta or fetal head tissues in ZIKV-117- or PBS-treated pregnant mice.
As described in Fig. 3, wild-type female mice were mated with wild-type sires and monitored for pregnancy. At E5.5, dams were treated with anti-Ifnar1 mAb and PBS or 250 μg of ZIKV-117. One day later (E6.5), dams were inoculated with 103 FFU of ZIKV-Dakar. Fetuses and placentas (n = 4 each) were collected on E13.5, homogenized, and tested for human IgG by ELISA. Human antibody in tissues was captured on ELISA plates coated with ZIKV E protein and detected using goat anti-human IgG (Fc-specific) antibody. The quantity of antibody was determined by comparison with a standard curve constructed using purified ZIKV-117 in a dilution series. Four replicate measurements were performed for each mouse tissue and the results were averaged. The graphs represent the mean + s.e.m. from 3 mice per group.
Extended Data Figure 5 Comparison of wild-type and LALA-mutated antibodies.
a, Binding to recombinant human FcγR1. The functional abrogation of the binding of the LALA variant IgG was confirmed in an ELISA binding assay with recombinant human FcγRI. ZIKV-117 wild-type bound to FcγRI, whereas the ZIKV-117 LALA antibody did not. Wild-type and LALA versions of another human mAb, CKV063, were used as controls. Binding to human FcγRI is one representative experiment of two, and error bars indicate s.e.m. of triplicate technical replicates. b, Neutralization assays. Wild-type ZIKV-117 and LALA antibodies exhibited equivalent neutralizing activity in vitro to each other and to the hybridoma-derived antibody. Neutralization assays are representative of two independent experiments completed in triplicate.
Extended Data Figure 6 In situ hybridization of Ifnar1+/− placenta after inoculation with ZIKV-Brazil and treatment with ZIKV-117.
As described in Fig. 3a, Ifnar1−/− female mice were mated with wild-type sires and monitored for pregnancy. At E5.5, dams were treated with 250 μg of either hCHK-152 isotype control or ZIKV-117. At E6.5, dams were inoculated with 103 FFU of ZIKV-Brazil. Collected placentas were fixed in 10% neutral buffered formalin at ambient temperature and embedded in paraffin. At least three placentas from different litters with the indicated treatments were sectioned for in situ hybridization staining using negative or ZIKV-specific RNA probes. Low (scale bar, 500 μm) and high (scale bar, 50 μm) power images are presented in sequence.
Rights and permissions
About this article

Cite this article
Sapparapu, G., Fernandez, E., Kose, N. et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540, 443–447 (2016). https://doi.org/10.1038/nature20564
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature20564
This article is cited by
-
A gossypol derivative effectively protects against Zika and dengue virus infection without toxicity
BMC Biology (2022)
-
Vaccination with a Zika virus envelope domain III protein induces neutralizing antibodies and partial protection against Asian genotype in immunocompetent mice
Tropical Medicine and Health (2022)
-
ZIKV-envelope proteins induce specific humoral and cellular immunity in distinct mice strains
Scientific Reports (2022)
-
The key amino acids of E protein involved in early flavivirus infection: viral entry
Virology Journal (2021)
-
ZIKV viral proteins and their roles in virus-host interactions
Science China Life Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
