
Abstract
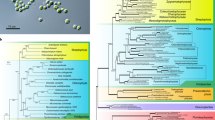
Brown algae (Phaeophyceae) are complex photosynthetic organisms with a very different evolutionary history to green plants, to which they are only distantly related1. These seaweeds are the dominant species in rocky coastal ecosystems and they exhibit many interesting adaptations to these, often harsh, environments. Brown algae are also one of only a small number of eukaryotic lineages that have evolved complex multicellularity (Fig. 1). We report the 214 million base pair (Mbp) genome sequence of the filamentous seaweed Ectocarpus siliculosus (Dillwyn) Lyngbye, a model organism for brown algae2,3,4,5, closely related to the kelps6,7 (Fig. 1). Genome features such as the presence of an extended set of light-harvesting and pigment biosynthesis genes and new metabolic processes such as halide metabolism help explain the ability of this organism to cope with the highly variable tidal environment. The evolution of multicellularity in this lineage is correlated with the presence of a rich array of signal transduction genes. Of particular interest is the presence of a family of receptor kinases, as the independent evolution of related molecules has been linked with the emergence of multicellularity in both the animal and green plant lineages. The Ectocarpus genome sequence represents an important step towards developing this organism as a model species, providing the possibility to combine genomic and genetic2 approaches to explore these and other4,5 aspects of brown algal biology further.

Here we define groups showing complex multicellularity as those that include macroscopic organisms with defined, recognizable morphologies and composed of multiple cell types. Coloured bars indicate the approximate, relative times of emergence of complex multicellularity in each lineage. The inset tree to the right indicates the relationship of Ectocarpus to selected brown algal genera. Kelps are represented in the tree by the genus Laminaria.
Similar content being viewed by others


Main
The 16,256 protein coding genes present in the 214 Mbp haploid male genome of E. siliculosus are rich in introns (seven per gene on average), have long 3′ untranslated regions (average size: 845 bp) and are often located very close to each other on the chromosome (29% of the intergenic regions between divergently transcribed genes are less than 400 bp long; Table 1 and Supplementary Information 2.1).
Repeated sequences, including DNA transposons, retrotransposons and helitrons, make up 22.7% of the Ectocarpus genome. Small RNAs mapped preferentially to transposons, indicating that they have a role in silencing these elements despite the absence of detectable levels of cytosine methylation in the genome (Supplementary Information 2.1). Sequencing also revealed the presence of an integrated copy of a large DNA virus, closely related to the Ectocarpus phaeovirus EsV-1 (ref. 8; Fig. 2a). Approximately 50% of individuals in natural Ectocarpus populations show symptoms of viral infection9,10 but the sequenced Ectocarpus strain Ec 32 has never been observed to produce virus particles and expression analysis showed that almost all of the viral genes were silent (Fig. 2b and Supplementary Information 2.1.17).

a, Representation of the linear and circular forms of the EsV-1 genome compared to the inserted viral genome. Genes on the upper and lower strands are above and below the line, respectively. A short region of the viral genome containing the putative integrase gene has been transposed to supercontig 0371. Gray parallelograms connect regions of high gene density that are also found in other phaeoviruses. These regions contain many of the genes that are thought to be important for the viral life cycle. Percent nucleotide identities between cognate regions in EsV-1 and in the integrated viral genome are indicated. Dashed line, algal DNA. ITRA and ITRA', inverted terminal repeats. b, Mean expression levels of the inserted viral genes: the graph shows the mean of the normalized expression values (4 replicates) ± s.d. of the genes that were included in the microarray experiments carried out in ref. 29. Expression data are shown for the control condition, but gene expression profiles were highly similar under stress conditions (not shown). Each bar represents the expression value for one coding sequence, the bars are in the same order as the corresponding genes along the supercontigs. Red bars correspond to virus genes, blue bars to host genes. Supercontigs 0062, 0052 and 0028 are adjacent on the genetic map, supercontig 0371 is part of another linkage group. The hybridization signals for 95% of the negative controls (median of four random probes on the same array) were between 19 and 59 (indicated by the two dotted lines).
The shallow waters of the intertidal region are an attractive habitat for marine, sedentary, photosynthetic organisms providing them with both a substratum and access to light. However, the shoreline is a also a hostile environment necessitating an ability to cope with tidal changes in light intensity, temperature, salinity and wave action, and with the biotic stresses characteristic of dense coastal ecosystems. Several features of the Ectocarpus genome indicate that this alga has evolved effective mechanisms for survival in this environment (Supplementary Information 2.2). For example, there is a large family of light harvesting complex (LHC) genes in Ectocarpus (53 loci, although some are probably pseudogenes), including a cluster of 11 genes with highest similarity to the LI818 family of light-stress related LHCs. The Ectocarpus genome is also predicted to encode a light-independent protochlorophyllide reductase (DPOR), allowing efficient synthesis of chlorophyll under dim light (Supplementary Information 2.2.2 and 2.2.3). Together these data indicate that Ectocarpus has a complex photosynthetic system that should enable it to adapt to an environment with highly variable light conditions. The high levels of phenolic compounds in brown algae are thought to protect against ultraviolet radiation, in a manner analogous to flavonoids in terrestrial plants11. Homologues of most of the terrestrial plant flavonoid pathway genes were found in Ectocarpus but these are completely absent from diatom or green algal genomes (Supplementary Information 2.2.9). The diverse complement of enzymes involved in the metabolism of reactive oxygen species (Supplementary Information 2.2.11) is also likely to represent an important adaptation to osmotic and light stresses.
In the Laminariales, the high concentration of apoplastic iodide is thought to be used in a new anti-oxidant system that, through the emission of iodine, has an impact on atmospheric chemistry12. Ectocarpus also accumulates halides, although to a significantly lower level than in kelps (Supplementary Information 2.2.10). This difference was reflected in the genome; only one vanadium-dependent bromoperoxidase was found in contrast to the large families of haloperoxidases in Laminaria digitata13. The Ectocarpus genome does, however, encode 21 putative dehalogenases and two haloalkane dehalogenases. These enzymes may serve to protect Ectocarpus against halogenated compounds produced by kelps as defence molecules12, allowing it to grow epiphytically on these organisms14,15.
The cell walls of brown algae contain unusual polysaccharides such as alginates and fucans16, with properties that are important both in terms of resistance to mechanical stresses and as protection from predators. Analysis of the Ectocarpus genome failed to detect homologues of many of the enzymes that are known, from other organisms, to have roles in alginate biosynthesis and in the remodelling of alginates, fucans and cellulose, indicating that brown algae have independently evolved enzymes to carry out many of these processes. However, a number of polysaccharide modifying enzymes, such as mannuronan C5 epimerases, sulphotransferases and sulphatases, were identified. These enzymes are likely to modulate physicochemical properties of the cell wall, influencing rigidity, ion exchange16 and resistance to abiotic stress.
Comparison of genomes from a broad range of organisms (Fig. 3) indicated that the major eukaryotic groups have retained distinct but overlapping sets of genes since their evolution from a common ancestor, with new gene families evolving independently in each lineage. On average, lineages that have given rise to multicellular organisms have lost fewer gene families and evolved more new gene families than unicellular lineages. However, we were not able to detect any significant, common trends, such as a tendency for the multicellular lineages to gain families belonging to particular functional (gene ontology) groups.

The number of gene families that were acquired (black) or lost (red) at each time point (grey circles) in the tree (Supplementary Information 1.15) was estimated using the Dollo parsimony principle. For each species, the number of orphans (genes that lacked homologues in the eukaryotic data set), the total number of gene families gained or lost and the overall gain (that is, total gain minus total loss) is indicated.
Analysis of the gene families that are predicted to have been gained by the Ectocarpus genome since divergence from the unicellular diatoms indicated a significant gain in ontology terms associated with protein kinase activities, and these genes include a particularly interesting family of membrane-spanning receptor kinases. Receptor kinases have been shown to have key roles in developmental processes such as differentiation and cellular patterning in both the animal and green plant lineages17. Animal tyrosine and green plant serine/threonine receptor kinases form two separate monophyletic clades, indicating that these two families evolved independently, and in both lineages the emergence of receptor kinases is thought to have been a key event in the evolution of multicellularity18,19. The Ectocarpus receptor kinases also form a monophyletic clade, discrete from those of animal and green plant receptor kinases, indicating that the brown algal family also evolved independently (Fig. 4). The evolution of membrane-spanning receptor kinases may, therefore, have been a key step in the evolution of complex multicellularity in at least three of the five groups that have attained this level of developmental sophistication. No orthologues of the Ectocarpus receptor kinase family were found in other stramenopile genomes, but a detailed analysis of two complete oomycete genome sequences identified a phylogenetically distinct family of receptor kinases (Fig. 4).

Protein maximum-likelihood tree generated using a multiple alignment of kinase domains from eukaryotic receptor kinases and related cytosolic kinases. Bootstrap values, when above 65%, are provided at the nodes for maximum-likelihood (first value) and neighbour-joining (second value) analyses.
The Ectocarpus genome contains a number of other genes that could have potentially had important roles in the development of multicellularity (see Supplementary Information 2.3; although it should be noted that the functions of these proteins will need to be confirmed experimentally). For example there are several additional membrane-localized proteins of interest, including three integrin-related proteins. Integrins have an important role in cell adhesion in animals20 but integrin genes are absent from all the previously sequenced stramenopile genomes. The Ectocarpus genome also encodes a large number of ion channels, compared to other stramenopile genomes. These include several channels that are likely to be involved in calcium signalling such as an inositol triphosphate/ryanodine type receptor (IP3R/RyR), four 4-domain voltage-gated calcium channels, and an expanded family of 18 transient receptor potential channels. Members of all these classes are found in animal genomes but are absent from the genomes of land plants21,22. No IP3R genes have been identified in the sequenced diatom and oomycete genomes, but the presence of an IP3R in Ectocarpus is consistent with the demonstration of ‘animal-like’ fast calcium waves and inositol-phosphate-induced calcium release in embryos of the brown alga Fucus serratus23,24.
The ion channels in the Ectocarpus genome illustrate how the evolutionary fates of eukaryotic lineages have probably depended not only on the evolution of new gene functions but also on the retention of genes already present in ancestral genomes. Along similar lines, there is evidence that, compared to unicellular organisms, multicellular organisms have tended to retain a more complete Rad51 family, which encodes DNA repair proteins including members with important roles during meiosis25. This is also the case in the stramenopiles, where Ectocarpus has a markedly more complete Rad51 gene family than the other sequenced members of the group (Supplementary Information 2.3.12). Ectocarpus also possesses a more extensive set of GTPase genes than other stramenopile genomes (Supplementary Information 2.3.7) and an analysis of transcription-associated proteins indicated that Ectocarpus and oomycete genomes have a broader range of transcription factor families than the unicellular diatoms (Supplementary Table 4).
Analysis of a large set of small RNA sequences allowed the identification of 26 microRNAs in Ectocarpus (Supplementary Table 17). This observation, together with the identification of microRNAs in three other eukaryotic groups, the archaeplastid, opisthokont and amoebozoan lineages26, indicates that these regulatory molecules were present from an early stage of eukaryotic evolution. Sixty-seven candidate target sites were identified for 12 of the 26 microRNAs. Interestingly, 75% of these target sequences occur in genes with leucine-rich repeat (LRR) domains (Supplementary Information 2.3.14). The LRR genes include many members of the ROCO (Roc GTPase plus COR (C-terminal of Roc) domain) family27 that are predicted to have evolved since the split from the diatoms. Taken together, these observations indicate that a significant proportion of the microRNAs identified may regulate recently evolved processes. This is interesting in the light of suggestions that microRNAs may have had a key role in the evolution of complex multicellularity in the animal lineage28.
Analysis of the Ectocarpus genome has revealed traces both of its ancient evolutionary past and of more recent events associated with the emergence of the brown algal lineage. The former include the diverse origins of the genes that make up the genome, many of which were acquired via endosymbiotic events (Supplementary Information 2.3.15), whereas the latter include the recent emergence of new gene families and the evolution of an unusual genome architecture, in terms both of gene structure and organization (Supplementary Information 2.1). It is likely that the evolution of complex multicellularity within brown algae depended on events spanning both timescales. The conservation of completeness and diversity within key gene families over the long term seems to have been as important as the more recent evolution of novel proteins, such as the brown algal receptor kinase family.
Methods Summary
Genome and cDNA sequencing were carried out using the Ectocarpus siliculosus strain Ec 32, which is a meiotic offspring of a field sporophyte collected in 1988 in San Juan de Marcona, Peru. The genome sequence was assembled using 2,233,253 and 903,939 paired, end-sequences from plasmid libraries with 3 and 10 kbp inserts respectively, plus 58,155 paired, end-sequence reads from a small-insert bacterial artificial chromosome library. Annotation was carried out using the EuGène program and optimized by manual correction of gene models and functional assignments. Sequencing of 91,041 cDNA reads, corresponding to six different cDNA libraries, and a whole genome tiling array analysis provided experimental confirmation of a large proportion of the transcribed part of the genome (Table 1). Small RNAs were characterized by generating 7,114,682 sequencing reads from two small RNA libraries on a Solexa Genome Analyser (Illumina). Analyses of the methylation state of genomic DNA and of specific transposon families were carried out using HPLC analysis of nucleotide methylation and McrBC digestion, respectively. Full information about the methodology used can be found in the Supplementary Information section.
Accession codes
Primary accessions
EMBL/GenBank/DDBJ
Gene Expression Omnibus
Data deposits
The annotated Ectocarpus genome sequence can be obtained through the EMBL Nucleotide Sequence Database (accession numbers CABU01000001–CABU01013533, FN647682–FN649242, FN649726–FN649760) and can be browsed at the Bogas website (http://bioinformatics.psb.ugent.be/webtools/bogas/). cDNA sequence data are available through accession numbers FP245546–FP312611 and small RNA sequences and tiling array data have been submitted to the GEO database (accession numbers ERA000209 and GSE19912, respectively). The Ectocarpus microRNAs have been submitted to miRBase (accession numbers esi-MIR3450–esi-MIR3469).
References
Yoon, H. S., Hackett, J. D., Ciniglia, C., Pinto, G. & Bhattacharya, D. A molecular timeline for the origin of photosynthetic eukaryotes. Mol. Biol. Evol. 21, 809–818 (2004)
Peters, A. F. et al. Life-cycle-generation-specific developmental processes are modified in the immediate upright mutant of the brown alga Ectocarpus siliculosus . Development 135, 1503–1512 (2008)
Peters, A. F., Marie, D., Scornet, D., Kloareg, B. & Cock, J. M. Proposal of Ectocarpus siliculosus (Ectocarpales, Phaeophyceae) as a model organism for brown algal genetics and genomics. J. Phycol. 40, 1079–1088 (2004)
Charrier, B. et al. Development and physiology of the brown alga Ectocarpus siliculosus: two centuries of research. New Phytol. 177, 319–332 (2008)
Coelho, S. M. et al. Complex life cycles of multicellular eukaryotes: new approaches based on the use of model organisms. Gene 406, 152–170 (2007)
Kawai, H., Hanyuda, T., Draisma, S. G. A. & Müller, D. G. Molecular phylogeny of Discosporangium mesarthrocarpum (Phaeophyceae) with a reinstatement of the order Discosporangiales. J. Phycol. 43, 186–194 (2007)
Phillips, N., Burrowes, R., Rousseau, F., de Reviers, B. & Saunders, G. W. Resolving evolutionary relationships among the brown algae using chloroplast and nuclear genes. J. Phycol. 44, 394–405 (2008)
Delaroque, N. et al. The complete DNA sequence of the Ectocarpus siliculosus virus EsV-1 genome. Virology 287, 112–132 (2001)
Dixon, N. M., Leadbeater, B. S. C. & Wood, K. R. Frequency of viral infection in a field population of Ectocarpus fasciculatus (Ectocarpales, Phaeophyceae). Phycologia 39, 258–263 (2000)
Müller, D. G. et al. Massive prevalence of viral DNA in Ectocarpus (Phaeophyceae, Ectocarpales) from two habitats in the North Atlantic and South Pacific. Bot. Mar. 43, 157–159 (2000)
Rozema, J. et al. The role of UV-B radiation in aquatic and terrestrial ecosystems–an experimental and functional analysis of the evolution of UV-absorbing compounds. J. Photochem. Photobiol. B 66, 2–12 (2002)
Küpper, F. C. et al. Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proc. Natl Acad. Sci. USA 105, 6954–6958 (2008)
Colin, C. et al. The brown algal kelp Laminaria digitata features distinct bromoperoxidase and iodoperoxidase activities. J. Biol. Chem. 278, 23545–23552 (2003)
Russell, G. Formation of an ectocarpoid epiflora on blades of Laminaria digitata . Mar. Ecol. Prog. Ser. 11, 181–187 (1983)
Russell, G. Parallel growth-patterns in algal epiphytes and Laminaria blades. Mar. Ecol. Prog. Ser. 13, 303–304 (1983)
Kloareg, B. & Quatrano, R. S. Structure of the cell walls of marine algae and ecophysiological functions of the matrix polysaccharides. Oceanogr. Mar. Biol. 26, 259–315 (1988)
De Smet, I., Voss, U., Jürgens, G. & Beeckman, T. Receptor-like kinases shape the plant. Nature Cell Biol. 11, 1166–1173 (2009)
Shiu, S. H. & Bleecker, A. B. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl Acad. Sci. USA 98, 10763–10768 (2001)
Cock, J. M., Vanoosthuyse, V. & Gaude, T. Receptor kinase signalling in plants and animals: distinct molecular systems with mechanistic similarities. Curr. Opin. Cell Biol. 14, 230–236 (2002)
Arnaout, M. A., Goodman, S. L. & Xiong, J.-P. Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 19, 495–507 (2007)
Nakayama, Y., Fujiu, K., Sokabe, M. & Yoshimura, K. Molecular and electrophysiological characterization of a mechanosensitive channel expressed in the chloroplasts of Chlamydomonas . Proc. Natl Acad. Sci. USA 104, 5883–5888 (2007)
Wheeler, G. L. & Brownlee, C. Ca2+ signalling in plants and green algae – changing channels. Trends Plant Sci. 13, 506–514 (2008)
Goddard, H., Manison, N., Tomos, D. & Brownlee, C. Elemental propagation of calcium signals in response-specific patterns determined by environmental stimulus strength. Proc. Natl Acad. Sci. USA 97, 1932–1937 (2000)
Coelho, S. M. et al. Spatiotemporal patterning of reactive oxygen production and Ca2+ wave propagation in Fucus rhizoid cells. Plant Cell 14, 2369–2381 (2002)
Lin, Z., Kong, H., Nei, M. & Ma, H. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl Acad. Sci. USA 103, 10328–10333 (2006)
Griffiths-Jones, S., Saini, H., van Dongen, S. & Enright, A. miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158 (2008)
Marín, I., van Egmond, W. N. & van Haastert, P. J. M. The Roco protein family: a functional perspective. FASEB J. 22, 3103–3110 (2008)
Peterson, K. J., Dietrich, M. R. & McPeek, M. A. MicroRNAs and metazoan macroevolution: insights into canalization, complexity, and the Cambrian explosion. Bioessays 31, 736–747 (2009)
Dittami, S. M. et al. Global expression analysis of the brown alga Ectocarpus siliculosus (Phaeophyceae) reveals large-scale reprogramming of the transcriptome in response to abiotic stress. Genome Biol. 10, R66 (2009)
Acknowledgements
We would like to thank Dieter G. Müller for his help and advice. The project was supported by the French GIS ‘Institut de la Génomique Marine’, the Centre National de Recherche Scientifique, the European Union network of excellence Marine Genomics Europe, the GIS Europôle Mer, the Inter-University Network for Fundamental Research (P6/25, BioMaGNet), the ‘Conseil Général’ of the Finistère department and the University Pierre and Marie Curie.
Author information
Authors and Affiliations
Contributions
J.M.C. coordinated genome analysis and manuscript preparation. P.W. and Y.V.d.P. coordinated genome assembly and centralized and enabled the annotation process, respectively. P.W. and Y.V.d.P. should be considered joint last authors. L.S. and P.R. implemented the automated annotation of the genome and made substantial contributions to genome annotation and analysis. D.S. developed and implemented protocols for library construction. L.S., P.R and D.S. should be considered joint second authors. All other authors are members of the genome sequencing consortium and contributed annotation, analyses or data to the genome project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Methods (1.1-1.21), Supplementary Notes comprising: Genome Structure and organization (2.1-2.18); Metabolism (2.2.1-2.2.13); Signalling and cell biology (2.3.1-2.3.16) and References. (PDF 1514 kb)
Supplementary Figures
This file contains Supplementary Figures 1-55 with legends. (PDF 9415 kb)
Supplementary Tables
This file contains Supplementary Tables 1-42. (PDF 1128 kb)
Supplementary Table 43
This table shows genes predicted to be derived from an endosymbiotic event involving a red alga. (XLS 1601 kb)
Supplementary Table 44
This table shows genes predicted to be derived from an endosymbiotic event involving a green alga. (XLS 677 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Cock, J., Sterck, L., Rouzé, P. et al. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465, 617–621 (2010). https://doi.org/10.1038/nature09016
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature09016
This article is cited by
-
The brown seaweed Ectocarpus
Nature Methods (2024)
-
The genomes of Vischeria oleaginous microalgae shed light on the molecular basis of hyper-accumulation of lipids
BMC Biology (2023)
-
Genome analysis of Parmales, the sister group of diatoms, reveals the evolutionary specialization of diatoms from phago-mixotrophs to photoautotrophs
Communications Biology (2023)
-
Iron uptake, transport and storage in marine brown algae
BioMetals (2023)
-
Glutathione S-Transferase (GST) Identified from Giant Kelp Macrocystis pyrifera Increases the Copper Tolerance of Synechococcus elongatus PCC 7942
Journal of Ocean University of China (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
