
Abstract
Lichen symbiosis is centered around a relationship between a fungus and a photosynthetic microbe, usually a green alga. In addition to their main photosynthetic partner (the photobiont), lichen symbioses can contain additional algae present in low abundance. The biology of these algae and the way they interact with the rest of lichen symbionts remains largely unknown. Here we present the first genome sequence of a non-photobiont lichen-associated alga. Coccomyxa viridis was unexpectedly found in 12% of publicly available lichen metagenomes. With few exceptions, members of the Coccomyxa viridis clade occur in lichens as non-photobionts, potentially growing in thalli endophytically. The 45.7 Mbp genome of C. viridis was assembled into 18 near chromosome-level contigs, making it one of the most contiguous genomic assemblies for any lichen-associated algae. Comparing the C. viridis genome to its close relatives revealed the presence of traits associated with the lichen lifestyle. The genome of C. viridis provides a new resource for exploring the evolution of the lichen symbiosis, and how symbiotic lifestyles shaped evolution in green algae.
Similar content being viewed by others

Introduction
The discovery of symbiosis started with lichens—complex symbiotic assemblages, in which symbiotic partners are tightly integrated into a single body (the thallus), which is often three dimensional and separated into tissue-like layers1. The classic definition of a lichen involved two, rarely three, partners: one fungus (the mycobiont), plus one microscopic green alga (the photobiont) and/or one cyanobacterium, which collectively constitute a lichen. This definition, however, has proven too simplistic, since many lichens contain microbial organisms in addition to the main partners, typically bacteria and yeasts2,3,4,5.
Studies on algae in lichens have also shown surprising diversity. Instead of one algal strain, as had been assumed before, some lichens contain two or more coexisting within the same thallus (reviewed by Muggia et al.6). Often, such co-occurring algae are closely related species of Trebouxia or another typical photobiont genus (for examples see7,8,9,10,11). Although direct evidence is still lacking, these algae are believed to reside within the algal layer together and occupy more or less the same niche in the symbiotic relationship. In other words, algal cells that were previously assumed to belong to one uniform photobiont, turned out to be two different, albeit closely related photobionts. However, this is not the only instance of algal diversity within lichens.
Compared to lichen photobionts, much less is known about other algae present in lichens. Coming from diverse groups and present in miniscule amounts, these ‘additional’ green algae have recently been termed the phycobiome12 They are mostly detected in two ways. First, by culturing them from a lichen thallus (e.g.,13,14,15,16). Sometimes this happens by accident, when researchers attempt to culture lichen photobionts17. Second, during metabarcoding surveys, in which, when reported, non-photobiont algae represent a small fraction of produced data (e.g.,18,19,20,21). These algae are sometimes assumed to grow epiphytically on lichens (e.g.,13,14), which is supported by studies comparing algal sequences from washed and unwashed lichen thalli18,22. However, much remains unknown about the non-photobiont lichen-associated algae, and the hypothesis that they partake in lichen symbioses cannot be ruled out.
Here, we examine one such alga, Coccomyxa viridis, and its distribution in lichen symbioses. We report the first sequenced genome of a non-photobiont lichen-associated alga, and compare it to genomes of closely related algae. We identify characteristics of its genome that are consistent with its lichen-associated lifestyle and discuss its potential role within the symbiosis.
Results
Non-photobiont Coccomyxa cultured from a Xanthoria lichen
A strain of green alga (Fig. 1a) was cultured from a thallus of the Xanthoria parietina lichen (Fig. 1b). While we originally expected to isolate Trebouxia, the main photobiont of Xanthoria23, it appeared to be overgrown by a different alga. Instead of globular cells with a large star-shaped and centrally-located chloroplast, as is the case for Trebouxia (Fig. 1a,c;24), the algal cells in our cultures were ellipsoidal with chloroplasts in the cell periphery (Fig. 1a).

Algae in Xanthoria parietina. (a) Micrographs of Coccomyxa viridis cultured from a X. parietina thallus. C. viridis cells are ellipsoid and have one or several chloroplasts located near cell exterior. For comparison, the lower track shows the main photobiont of X. parietina, Trebouxia. Trebouxia cells are globular and contain one centrally located chloroplast, often star-shaped or lobed. In both cultures, cell walls were stained with the Calcofluor White (CFW) stain. Scale bar = 5 μm. (b) Thallus of X. parietina growing on a tree branch; photo courtesy of Phil Robinson. (c) Cross-section through a X. parietina thallus, showing internal structure with four layers: uc = upper cortex (formed primarily by mycobiont hyphae embedded in an extracellular matrix), al = algal layer (mycobiont hyphae and photobiont cells), me = medulla (loosely arranged mycobiont hyphae), lc = lower cortex (mycobiont hyphae embedded in an extracellular matrix). The arrow points to a cell of Trebouxia residing in the algal layer. Scale bar = 50 μm.
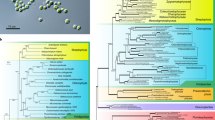
By constructing a phylogenomic tree with publicly available Trebouxiophyceae genomes, we identified our alga as a part of the larger Elliptochloris clade (Fig. 2a). A phylogeny based on the internal transcribed spacer (ITS) placed our strain in the Coccomyxa viridis clade (Fig. 2b).

Maximum-likelihood phylogenetic trees showing the taxonomic placement of the studied strain (highlighted in red). (a) Phylogenomic tree based on 196 single-copy orthologs. The green rectangle highlights the Elliptochloris-clade. (b) Phylogeny based on ITS sequences. The blue rectangle highlights the C. viridis clade.
Coccomyxa viridis detected in 12% of lichen metagenomes
Apart from identifying the target strain, our phylogenetic analysis showed that the majority of its known close relatives are also lichen-associated. The C. viridis clade contained numerous lichen-associated strains, which can be broken into two categories (Fig. 2b). Firstly, five Coccomyxa strains that are main photobionts of their lichens: three different Micarea lichens and Schizoxylon albescens. Of the lichen photobionts included in this analysis, C. viridis represented only a minority, as most of them were recovered in a different part of the tree, in the C. subellipsoidea and C.simplex/C. solorinae clades. Secondly and more notably, 11 non-photobiont alga—i.e. algae cultured from lichens that do not have Coccomyxa as their main photobiont. This begged the question of how widespread C. viridis is in lichens.
We screened 438 publicly available lichen metagenomes for the presence of Coccomyxa ITS. The screened metagenomes originated from 14 studies and were collected from different substrates and geographic locations. In total, we found 84 Coccomyxa sequences, which we used to construct a phylogenetic tree along with sequences from the published literature (Fig. 3a). While not all of them came from the C. viridis clade, the majority (82%) were identified as C. viridis. C. viridis was present in 53 lichen metagenomes (12% of the screened metagenomes), coming from different lichen groups, collectors, and geographic locations (Fig. 3b-g, Supplementary Table S1 online). Nearly all of these algae (88%) are likely not the main photobionts of their respective lichens, as they were detected in lichen symbioses known to have non-Coccomyxa photobionts (Supplementary Table S1 online).

C. viridis presence in publicly available lichen metagenomic data. (a) Phylogenetic tree of Coccomyxa ITS sequences. Pink dots represent sequences pulled from lichen metagenomic data. Blue represents C. viridis clade and C. viridis sequences from the literature. (b) Map showing geographic locations of each lichen sample, in which we detected C. viridis by screening metagenomic data produced from this sample. All these samples were collected in North America and Europe, however the real distribution of C. viridis could be broader, given that existing metagenomic data on lichens is geographically biased towards these two continents. (c) Presence of C. viridis across lichen taxonomic groups. The tree represents the phylogeny of lichen mycobionts modified from Tagirdzhanova et al.69 and Díaz-Escandón et al.73; only taxa included in the metagenomic screening are shown. Green dots show taxonomic groups for which C. viridis was detected, with the prevalence ratios shown to the right. (d-g) Examples of lichen symbioses containing C. viridis; photos courtesy of Jason Hollinger. d. Chrysothrix xanthina (Arthoniales, Arthoniomycetes). (e) Dibaeis baeomyces (Pertusariales, Lecanoromycetes). (f) Cladonia ochrochlora (Lecanorales, Lecanoromycetes). (g) Solorina crocea (Peltigerales, Lecanoromycetes).
The lichens that contained C. viridis originated from different substrates and included a wide variety of lichen taxonomic groups from classes Lecanoromycetes, Eurotiomycetes, Dothideomycetes, and Arthoniomycetes (Supplementary Table S1 online). The majority of these lichens possess green algae at their main photobionts, with only two exceptions that associate with cyanobacteria. Most commonly, C. viridis occurred in lichens that have Trebouxia as the main photobiont.
Is C. viridis external or internal to the symbiosis?
We washed eight samples of the X. parietina lichen and screened the resulting samples via PCR. After both gentle and aggressive washing, 75% of thalli still contained detectable C. viridis DNA (Fig. 4, Supplementary Fig. S1 online). In contrast, only one wash water sample had traces of C. viridis. Generic algal primers yielded Trebouxia sequences for all samples, confirming that Trebouxia and not C. viridis is the main photobiont of these lichens (Supplementary Table S2 online).

PCR-based screening for the C. viridis presence in washed lichen samples. Eight X. parietina thalli were used, of which a half were washed more gently in water, and each produced two DNA extractions: one from wash water and one from the washed thallus. The other half were washed more aggressively in ethanol and bleach; for those samples we only extracted DNA from the washed thallus. The top panel shows screening results for C. viridis-specific primers. Dark-green circles represent DNA extractions containing C. viridis DNA. Phylogenetic tree confirming the taxonomic assignment of C. viridis sequences is shown in Supplementary Fig. S1 online. The bottom panel shows screening results for generic algal rbcL primers. Light-green circles represent DNA extractions that yielded sequences of Trebouxia; white circles represent DNA extractions that did not yield a usable sequence.
Near chromosome-level genome assembly of C. viridis compared with its relatives
Assembly of the C. viridis nuclear genome amounted to 45.7 Mbp and 18 contigs (Fig. 5a,b), which is close to the existing chromosome-level genome assemblies from the Coccomyxa genus: a 50.9 Mbp genome of a non-lichen associated C. viridis25 and a 48.8 Mbp genome of C. subellipsoidea26. In ten contigs, we detected telomeric repeats TTTAGGG, which are typical in green algae27. Two contigs, cviridis_6 and cviridis_13, had telomeric sequences on both ends and likely represent complete chromosomes. The genome is estimated 97.5% complete according to BUSCO estimates and has a duplication rate of 0.2% (Fig. 5c). De novo annotation of the nuclear genome produced 11,248 gene models.

Genome of C. viridis. (a) Plot showing GC-content and length of the 18 contigs comprising the C. viridis nuclear genome. Red stripes show telomeric repeats. (b) Snailplot showing basic assembly statistics for the C. viridis nuclear genome. The gray bars show cumulative length of the assembly, with the red line showing the longest contigs. The orange and yellow lines represent N50 and N90 respectively. The plot design is based on Challis et al.74. (c) Genome completeness scores as estimated by BUSCO (chlorophyta_odb10 database). (d) Gene map of the mitochondrial genome. The inner circle represents GC-content. The genes are mapped on the outer circles, with genes inside the circle transcribed clockwise, and genes outside the circle transcribed counterclockwise. (e) Gene map of the plastid genome.
Plastid and mitochondrial genomes were assembled in a single circular contig each (Fig. 5d,e). The sizes of organelle genomes—64 kbp for the mitochondria and 210 kbp for the plastid—are similar to that of C. subellipsoidea, which has 65 kbp and 175 kbp respectively28. Both Coccomyxa species have organelle genomes that are uncommonly large for green algae28. While C. subellipsoidea is also reported to have an unusually high (> 50%) GC content in its organelle genomes28, our strain of C. viridis had a lower GC content of 42% and 40% respectively.
Our de-novo annotation of the C. viridis genome yielded 11,248 gene models and 11,202 protein records. In comparison to other published genomes of Coccomyxa algae, C. viridis has a slightly smaller genome, but a larger predicted proteome (the predicted proteomes of C. subellipsoidea and C. pringsheimii included 10,921 and 10,022 proteins respectively29; Fig. 6a). The types and number of secondary metabolism gene clusters in C. viridis were similar to the free-living C. subellipsoidea (Supplementary Table S3 online). In our functional annotations, we focused on the gene families identified by Puginier et al.29 and Armaleo et al.30 as connected with lichenization in green algae. Compared to other Coccomyxa species, C. viridis genome encoded comparable number of aquaporins, catalases, and domains similar to tryptophan-rich sensory protein/mitochondrial benzodiazepine receptor (TspO/MBR) (Fig. 6b)—groups of genes involved in stress response29. Unlike other studied Coccomyxa species, C. viridis genome did not encode any proteins from the Glycoside Hydrolase (GH) 8 family (as confirmed by both IntrePro and CAZy annotations)—a diverse family of hydrolases that includes licheninases, cellulases, chitosanases, and others. However, it encoded one protein from the GH16 family, which also contains licheninases. Most notably, C. viridis genome encoded several nitrile hydratases, which are typical in lichen photobionts29, yet were missing from a lichen photobiont C. pringsheimii (Fig. 6b). The signal transduction component in C. viridis is largely comparable to its relatives (Supplementary Fig. S2 online). However, the protein kinase family (IPR000719) appears expanded in C. viridis (Supplementary Table S4 online), reminiscent of similar expansions in the lichen photobiont Asterochloris30.

Comparative genomics analysis of C. viridis (highlighted in red) and other Trebouxiophyceae genomes. Information for the genomes other than C. viridis is taken from Puginier et al.29. (a) Basic genome statistics plotted by taxonomic group; the circles next to the species name represent the ecology of each strain. (b) Presence of InterProScan gene functional families across Trebouxiophyceae genomes. Here we show only functional families highlighted by Puginier et al.29 as potentially relevant to lichenization in green algae. The size of the bubbles represent the number of genes assigned to each family in a given genome.
Discussion
Here, we present evidence that green alga Coccomyxa viridis is widespread in lichens as a minor component present in addition to the main photobiont. C. viridis has been reported before from lichens with various non-Coccomyxa photobionts in several isolated reports13,14,16,20,21,31. Species from the C. viridis clade have been independently cultured from several lichen symbioses13,14,16. In addition, several amplicon metabarcoding studies of lichen algae reported small numbers of reads assigned to C. viridis20,21,31. Now, these reports are confirmed by our systematic screening of lichen metagenomic data. We detected C. viridis in one eighth of analyzed metagenomes. The lichen symbioses shown to contain C. viridis are quite diverse and the symbionts include representatives of both main groups of lichen photobionts (green algae and cyanobacteria) and several classes of mycobionts. Combined with reports of other non-photobiont algae frequent in lichens12,15, this finding raises questions about the place these algae occupy in the lichen symbiosis.
C. viridis comes from a genus that includes many symbiotic algae and, among others, lichen photobionts32. Most Coccomyxa photobionts come from one of the two clades: C. subellipsoidea and C. simplex/C. solorinae, which led to the hypothesis that lichenization happened in Coccomyxa twice33. Here, we show the C. viridis clade as a possible third independent origin of the lichen-associated lifestyle, which, however, differs from the other two. In both the C. subellipsoidea and C. simplex/C. solorinae clades, nearly all lichen-associated algae are photobionts. Our metagenomic screening, combined with the literature data, yielded only three occurrences of C. subellipsoidea and C. simplex/C. solorinae in lichens with non-Coccomyxa photobionts (Fig. 3a, Supplementary Table S1 online). In contrast, the majority of algae from the C. viridis clade were isolated from lichens with non-Coccomyxa photobionts, which suggests that they are lichen-associated non-photobiont algae. Several exceptions exist, as the C. viridis clade includes photobionts of several Micarea lichens34 and the photobiont of Schizoxylon albescens, an unusual lichen whose mycobiont is optionally lichenized and can occur as a non-symbiotic saprotroph35, plus a few strains with non-lichen ecologies, including a mussel parasite36. Overall, the fact that C. viridis can occur in lichens as either a photobiont or a non-photobiont is consistent with prior reports showing ‘additional’ algae in lichen thalli to be photobionts of unrelated lichens22. However, our results suggest that C. viridis, unlike other lichen-associated Coccomyxa species, primarily occurs in lichens as a non-photobiont.
How tightly is C. viridis associated with lichen symbioses? From the existing data we cannot determine how frequently C. viridis occurs outside of lichens, and therefore we cannot exclude the chance that C. viridis is a cosmopolitan alga so common that its presence in lichens is a mere coincidence. At the same time, the majority of existing C. viridis isolates originate from lichen material, with non-lichen ecology being comparatively rare. This, combined with the fact that C. viridis has been found in a wide variety of lichens from different substrates and continents, suggests some degree of association with lichens. Based on the available evidence, we hypothesize that C. viridis can exist both as a free-living and lichen-associates alga, but more commonly occurs in a lichen context, as is the case with other lichen algae, including the most common lichen photobiont Trebouxia22.
The newly sequenced genome of C. viridis is the first genome of a non-photobiont lichen-associated alga and one of the first near chromosome-level assemblies of any lichen-associated algae. It is also the fourth genome from Coccomyxa, in addition to free-living strains of C. subellipsoidea26 and C. viridis25 and the lichen photobiont C. pringsheimii (part of the C. simplex/C. solorinae clade)29. By comparing the three available genomes coming from different clades and different lifestyles, we showed that they share basic genomic characteristics (the fourth genome belonging to the free-living C. viridis has not been released at the time of submission). At the same time, our results suggest that C. viridis might exhibit more traits associated with lichenization compared to others, as demonstrated by a slight expansion of the kinase family and the presence of nitrile hydratases.
What is the nature of its relationship between non-photobiont algae such as C. viridis and the rest of lichen symbionts? While it is possible that non-photobiont algae only treat lichens as a substrate to attach to, they can potentially reap other benefits. For lichen photobionts, participation in the symbiosis is hypothesized to bring numerous rewards: protection from herbivory, access to nitrogen, and a better hydration regime (reviewed in37). The extent to which non-photobiont algae have access to the same benefits might depend on whether they grow epiphytically on the surface of lichen thalli, or in the thallus interior. Our screening of washed lichen samples suggests that C. viridis can be endophytic, however more evidence is needed to prove this conclusively. Conversely, other lichen symbionts might benefit from the non-photobiont algae. While carbohydrates produced by a small number of C. viridis cells are unlikely to significantly alter the carbon budget of the lichen, the presence of a diverse set of algae could facilitate photobiont-switching thereby increasing plasticity of the symbiosis as a whole22.
This study began with an accident. Our initial culture of the photobiont of a Xanthoria lichen was overgrown by C. viridis. Perhaps not completely coincidentally, the first sequenced genome of Coccomyxa, C. subellipsoidea, was also produced by accident in a project aimed at a different alga26. Relatively fast growth, observed for some non-photobiont lichen-associated algae17, and their frequent presence make C. viridis contamination a likely problem in studies involving culturing of lichen symbionts. At the same time, C. viridis and other frequently discarded and understudied members of lichen microbiota might yet shed light on the evolution of lichen symbiosis.
Currently, we know much less about the biology and the evolution of lichen-associated algae, compared to the lichen-associated fungi. This begins to change with a recent study pioneering comparative genomics of free-living algae and lichen photobionts29. We believe it can be beneficial to include non-photobiont lichen-associated algae into similar studies in the future, which will now be possible with the high-quality genome of C. viridis we provide. As we accumulate more information on the ecology of individual algal species and in what, if any, ways they engage in lichen symbioses, we will be able to chart the evolution of lichenization in green algae.
Methods
Culturing
The alga was cultured from a thallus of Xanthoria parietina lichen kindly provided by Prof. Paul Dyer, University of Nottingham, UK. The thallus was collected in the Peak District, UK. The photobiont was isolated from the thallus as previously described38,39. The culture was routinely grown in liquid Bold’s Mineral Medium (BMM) on a 12-h night/day light cycle.
Genome sequencing and assembly
DNA was extracted from 34 mg of dry weight of algal culture, which was snap-frozen, homogenized with a Geno/Grinder homogenizer (SPEX SamplePrep, Metuchen NJ, USA) at 1300 rpm for 1 min, and extracted with the NucleoBond High Molecular Weight DNA Kit (Macherey–Nagel, Düren, Germany). The extraction yielded 16.5 μg of high-molecular weight DNA, which was used for long-read sequencing. Short fragments were removed using Circulomics Short Read Eliminator Kit (Pacific Biosciences, Menlo Park CA, USA) with 25 kbp cut-off. A sequencing library was prepared using Native Barcoding Kit 96 V14 (Oxford Nanopore Technologies, Oxford, UK). The library was sequenced on a PromethION Flow Cell FLO-PRO114M (Oxford Nanopore Technologies, Oxford, UK) to 25 Gbp of data.
Basecalling was carried out using the ‘duplex’ method. Dorado v0.2.1 (Oxford Nanopore Technologies, Oxford, UK) was used for basecalling and Duplex tools v0.3.1 was used to identify duplex pairs. Contigs were de novo assembled with Flye v2.9-b178040 with ‘overlap 10 K, error rate 0.005, no-alt-contigs’ flags. The assembly was polished based on long reads using Medaka v1.7.2 (Oxford Nanopore Technologies, Oxford, UK). Long-read sequencing and assembly were performed by Future Genomics (Leiden, Netherlands).
In addition, we used the same DNA extraction to produce short read data. DNA was sent to Novogene UK (Cambridge, UK) and sequenced on an Illumina NovaSeq 6000 platform to 2 Gbp of PE150 data. The resulting short-read data were used to polish the long-read assembly with Pilon v1.2341.
Transcriptomic sequencing
We generated transcriptomic data to be used for training during the annotation of the genome. Algal culture was transferred from the liquid stock and plated on petri dishes with 99:1 BMM:MEYE culture medium. The cultures were harvested 2, 9, 21, and 42 days post inoculation, with three replicates for each time point. We snap-froze the harvested material in liquid nitrogen and extracted RNA using the RNeasy Plant Mini Kit (QIAGENE, Hilden, Germany). The RNA was sent to Novogene UK (Cambridge, UK) and sequenced on an Illumina HiSeq 2500 platform to PE150 data.
Genome annotation
Since our initial BLASTx search against NCBI-nr showed our genomic assembly to contain bacterial sequences, we used a metagenomic binning approach to filter out contamination. We aligned Illumina reads against the assembly using Bowtie242 and used the resulting bam file to bin the assembly with MetaBAT243. Next, we used the BLASTx search to select the bin that corresponded to the target algal genome. We confirmed the genome quality with BUSCO544, using the chlorophyta_odb10 database. To detect telomeric repeats, we used the script from Hiltunen et al.45 with ‘CCCTAAA’ as a query. To detect contigs representing organelle genomes, we used the results from the same BLASTx search.
Gene prediction and functional annotation of the nuclear genome was done using the Funannotate pipeline v1.8.1546. We masked repeated elements in the assembly using Tantan47 and generated gene prediction parameters using the ‘funannotate train’ command with RNA-seq data used for training. Gene prediction was performed using the ‘funannotate predict’ command, which performed ab initio prediction with Augustus v3.3.248, CodingQuarry v2.049, GlimmerHMM v3.0.450, and SNAP 2006-07-2851. Consensus models were created using EVidence Modeler v1.1.152. tRNA were predicted with tRNAscan-SE v2.0.953. Finally, functional annotation was done with the ‘funannotate annotate’ command. There, we assigned the gene models with putative functions based on the HMMER v3.3.2 and diamond v2.1.654 searches against several databases: PFAM v35.055, UniProt DB v2023_0156, MEROPS v12.057, dbCAN v11.058, and BUSCO chlorophyta_odb1044. We annotated the gene models with InterPro domains using InterProScan v5.42-78.059. To annotate secondary metabolism gene clusters, we used the antiSMASH v7.0.1 webserver60 in the fungal mode following O’Neill61. In order to compare our genome assembly and annotation to genomes of closely related alga, we used data from Puginier et al.29 and Armaleo et al.30.
Organelle genomes were annotated separately. We extracted contigs identified as organelle genomes from our initial assembly and predicted genes using MFannot62 and GeSeq63. To aid the annotation, we aligned RNA-seq data against the contigs identified as mitochondrial and plastid genomes using STAR v2.5.4b64. To finalize the annotations, we manually combined the outputs of the two tools and cross-referenced it against the RNA-seq alignment. The annotations were visualized using the OGDraw webserver65.
Phylogenetic analyses
To provide a taxonomic identification to the sequenced genome, we first built a phylogenomic tree using 10 reference genomes and transcriptomes from Trebouxiophyceae with Chlamydomonas eustigma as an outgroup (Supplementary Table S5 online). We identified chlorophyta_odb10 BUSCO single-copy orthologs shared by all genomes and transcriptomes, which amounted to 196 loci. Next, we created a single concatenated alignment using MAFFT v7.27166 and trimmed it with trimAL v1.267 to remove positions present in < 70% of organisms. Finally, we computed a phylogeny with RAxML v8.2.1268, using PROTGAMMAAUTO model. To provide a better taxonomic resolution, we created a tree based on the ITS region (ITS1, 5.8S ribosomal RNA gene, ITS2). We included 77 reference ITS sequences from Coccomyxa and Elliptochloris (Supplementary Table S6 online). The tree was constructed as described above.
Screening of publicly available metagenomic data
We searched for Coccomyxa viridis ITS in the 438 metagenomic assemblies from Tagirdzhanova et al.69. The metagenomes were sourced from NCBI and originated from 12 different studies, and 377 lichen symbioses (Supplementary Table S7 online). The procedure for metagenomic assembly is described in Tagirdzhanova et al.69. Briefly, each metagenomic dataset was filtered to remove human contamination, clipped to remove adapters, and assembled separately with metaSPAdes. To screen the metagenomic assemblies, we used a BLASTn search with the e-value cut-off of 1e-65. As a query, we used the ITS region (ITS1, 5.8S ribosomal RNA gene, ITS2) pulled from the genome assembly. Extracted hits were combined with the ITS reference sequences (Supplementary Table S2 online), aligned as described above, and used to construct a phylogeny using IQ-TREE v2.2.2.270 with 10,000 rapid bootstraps and the TIM2 + F + I + R3 substitution model.
Screening of lichen thalli
To determine if Coccomyxa viridis is external or internal to lichen thalli, we screened eight thalli of Xanthoria parietina lichen, in part following Moya et al.18. The lichen samples were collected in Norwich Research Park (Norwich, UK; 52.623133°N, 1.221621°E) from tree bark. We separated a 1 cm2 fragment of each thallus from its substrate taking care to remove all visible fragments of bark, moss, or other contaminants.
Four fragments were subjected to ‘soft’ washing. We soaked them in filter-sterilized water for 10 min, then vortexed for 5 min at 600 rpm. Next, we brushed the upper surface of thallus fragments with a soft paintbrush, following Yoshimura et al.71 and washed it in a jet of deionized water. Each thallus fragment yielded two samples: (1) washed lichen fragment and (2) wash water sample. We centrifuged the wash water samples at 4000 rpm for 5 min to obtain cell pellets. Next, we dried both washed lichen fragments and wash-water pellets at 65 °C and extracted DNA with the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany).
The second half of fragments were washed more aggressively. We followed the protocol from U’ren et al.72 for surface-sterilization of lichen thalli. First, we washed each fragment in a jet of deionized water. Next, we soaked and vortexed fragments at 600 rpm in three solutions: 95% ethanol for 10 s, 0.525% NaOCl for 2 min, and 75% ethanol for 2 min. Finally, the thalli were washed in a jet of deionized water for 2 min. We extracted DNA from the thalli as described above.
We screened the DNA extractions using two pairs of primers: specific for Coccomyxa viridis ITS, and general green algal primers for the large subunit of ribulose bisphosphate carboxylase (rbcL; Supplementary Table S8 online). PCR reactions were performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs, Ipswich MA, USA). For ITS we used the following conditions: 98 °C for 5 min, followed by 50 cycles of 98 °C for 30 s, 70 °C for 30 s, and 72 °C for 30 s, followed by the final extension step of 72 °C for 7 min. For rbcL we used the following conditions: 98 °C for 5 min, followed by 35 cycles of 98 °C for 1 min, 57 °C for 1 min, and 72 °C for 1 min, followed by the final extension step of 72 °C for 7 min. PCR reactions were gel-extracted using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison WI, USA) and sequenced by GENEWIZ (Leipzig, Germany). We used the resulting sequences to build a phylogeny with reference sequences as described above.
Microscopy
We visualized the culture of C. viridis and, for comparison, a culture of a Trebouxia photobiont isolated from a X. parietina thallus (the thallus was collected in Norwich Research Park; Trebouxia was isolated as described above). Algal samples were stained in Calcofluor White Stain (Sigma-Aldrich, Burlington MA, USA) for three minutes. Confocal microscopy was performed using a Leica SP8 laser confocal microscope with excitation wavelength 405 nm and emission wavelengths 410–430 nm for calcofluor white and 650–730 nm for the chlorophyll autofluorescence. In addition, we performed bright-field imaging of a cross-section through a X. parietina thallus using a Leica DM5500b microscope. Images were analyzed with Leica software and Fiji.
Data availability
The annotated genome assembly of C. viridis will be available at NCBI (Study Accession PRJEB65893). All code associated with the analysis along with details on the usage of bioinformatics tools is available on GitHub (https://github.com/metalichen/2023_Coccomyxa_viridis_genome).
References
Honegger, R. The lichen symbiosis: What is so spectacular about it?. Lichenologist 30, 193–212 (1998).
Grube, M. & Berg, G. Microbial consortia of bacteria and fungi with focus on the lichen symbiosis. Fung. Biol. Rev. 23, 72–85 (2009).
Spribille, T. et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 353, 488–492 (2016).
Grimm, M. et al. The lichens’ microbiota, still a mystery?. Front. Microbiol. 12, 839 (2021).
Scharnagl, K., Tagirdzhanova, G. & Talbot, N. J. The coming golden age for lichen biology. Curr. Biol. 33, R512–R518 (2023).
Muggia, L., Leavitt, S. & Barreno, E. The hidden diversity of lichenised Trebouxiophyceae (Chlorophyta). Phycologia 57, 503–524 (2018).
Casano, L. M. et al. Two Trebouxia algae with different physiological performances are ever-present in lichen thalli of Ramalina farinacea. Coexistence versus competition?. Environ. Microbiol. 13, 806–818 (2011).
Voytsekhovich, A. & Beck, A. Lichen photobionts of the rocky outcrops of Karadag massif (Crimean Peninsula). Symbiosis 68, 9–24 (2016).
Onuț-Brännström, I. et al. Sharing of photobionts in sympatric populations of Thamnolia and Cetraria lichens: Evidence from high-throughput sequencing. Sci. Rep. 8, 4406 (2018).
Vančurová, L., Muggia, L., Peksa, O., Řídká, T. & Škaloud, P. The complexity of symbiotic interactions influences the ecological amplitude of the host: A case study in Stereocaulon (lichenized Ascomycota). Mol. Ecol. 27, 3016–3033 (2018).
De Carolis, R. et al. Photobiont diversity in lichen symbioses from extreme environments. Front. Microbiol. 13, 809804 (2022).
Chiva, S., Moya, P. & Barreno, E. Lichen phycobiomes as source of biodiversity for microalgae of the Stichococcus-like genera. Biologia 78, 389–397 (2023).
Cao, S. et al. Coccomyxa antarctica sp. nov. from the Antarctic lichen Usnea aurantiacoatra. PhytoKeys 98, 107 (2018).
Cao, S. et al. Coccomyxa greatwallensis sp. nov. (Trebouxiophyceae, Chlorophyta), a lichen epiphytic alga from Fildes Peninsula, Antarctica. PhytoKeys 98, 39 (2018).
Chiva, S., Dumitru, C., Bordenave, C. D. & Barreno, E. Watanabea green microalgae (Trebouxiophyceae) inhabiting lichen holobiomes: Watanabea lichenicola sp. nova. Phycolog. Res. 69, 226–236 (2021).
Miral, A. et al. Microbial community associated with the crustose lichen Rhizocarpon geographicum L. (DC) living on oceanic seashore: A large source of diversity revealed by using multiple isolation methods. Environ. Microbiol. Rep. 14, 856–872 (2022).
Ahmadjian, V. A guide to the algae occurring as lichen symbionts: Isolation, culture, cultural physiology, and identification. Phycologia 6, 127–160 (1967).
Moya, P., Molins, A., Martínez-Alberola, F., Muggia, L. & Barreno, E. Unexpected associated microalgal diversity in the lichen Ramalina farinacea is uncovered by pyrosequencing analyses. PLoS One 12, e0175091 (2017).
Vančurová, L. et al. Symbiosis between river and dry lands: Phycobiont dynamics on river gravel bars. Algal Res. 51, 102062 (2020).
Molins, A., Moya, P., Muggia, L. & Barreno, E. Thallus growth stage and geographic origin shape microalgal diversity in Ramalina farinacea lichen holobionts. J. Phycol. 57, 975–987 (2021).
Xu, H., Wang, L., Feng, X. & Gong, X. Core taxa and photobiont-microbial interaction within the lichen Heterodermia obscurata (Physcsiaceae, Heterodermia). Symbiosis 86, 187–204 (2022).
Muggia, L. et al. The symbiotic playground of lichen thalli: A highly flexible photobiont association in rock-inhabiting lichens. FEMS Microbiol. Ecol. 85, 313–323 (2013).
Wyczanska, M., Wacker, K., Dyer, P. S. & Werth, S. Local-scale panmixia in the lichenized fungus Xanthoria parietina contrasts with substantial genetic structure in its Trebouxia photobionts. Lichenologist 55, 69–79 (2023).
Bordenave, C. D. et al. Chloroplast morphology and pyrenoid ultrastructural analyses reappraise the diversity of the lichen phycobiont genus Trebouxia (Chlorophyta). Algal Res. 61, 102561 (2022).
Kraege, A. et al. High quality genome assembly and annotation (v1) of the eukaryotic terrestrial microalga Coccomyxa viridis SAG 216–4. Biorxiv. https://doi.org/10.1101/2023.07.11.548521v1 (2023).
Blanc, G. et al. The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biol. 13, 1–12 (2012).
Fulnečková, J. et al. Dynamic evolution of telomeric sequences in the green algal order Chlamydomonadales. Genome Biol. Evol. 4, 248–264 (2012).
Smith, D. R. et al. The GC-rich mitochondrial and plastid genomes of the green alga Coccomyxa give insight into the evolution of organelle DNA nucleotide landscape. PLoS ONE 6, e23624 (2011).
Puginier, C. et al. Phylogenomics reveals the evolutionary origin of lichenization in chlorophyte algae. Biorxiv. https://doi.org/10.1101/2022.01.06.475074v2 (2022).
Armaleo, D. et al. The lichen symbiosis re-viewed through the genomes of Cladonia grayi and its algal partner Asterochloris glomerata. BMC Genomics 20, 1–33 (2019).
Faluaburu, M. S., Nakai, R., Imura, S. & Naganuma, T. Phylotypic characterization of mycobionts and photobionts of rock tripe lichen in East Antarctica. Microorganisms 7, 203 (2019).
Gustavs, L., Schiefelbein, U., Darienko, T., & Pröschold, T. Symbioses of the green algal genera Coccomyxa and Elliptochloris (Trebouxiophyceae, Chlorophyta). in Algal and Cyanobacteria Symbioses (eds. Grube, M., Seckbach, J., Muggia, L.) 169–208 (2017).
Pardo-De la Hoz, C. J. et al. Contrasting symbiotic patterns in two closely related lineages of trimembered lichens of the genus Peltigera. Front. Microbiol. 9, 2770 (2018).
Yahr, R., Florence, A., Skaloud, P. & Voytsekhovich, A. Molecular and morphological diversity in photobionts associated with Micarea s. str. (Lecanorales, Ascomycota). Lichenologist 47, 403 (2015).
Muggia, L., Baloch, E., Stabentheiner, E., Grube, M. & Wedin, M. Photobiont association and genetic diversity of the optionally lichenized fungus Schizoxylon albescens. FEMS Microbiol. Ecol. 75, 255–272 (2011).
Rodríguez, F. et al. Phylogenetic and morphological characterisation of the green algae infesting blue mussel Mytilus edulis in the North and South Atlantic oceans. Dis. Aquat. Org. 81, 231–240 (2008).
Spribille, T., Resl, P., Stanton, D. E. & Tagirdzhanova, G. Evolutionary biology of lichen symbioses. New Phytol. 234, 1566–1582 (2022).
Ahmadjian, V. Methods of isolating and culturing lichen symbionts and thalli. in The lichens (eds. Ahmadjian, V., Hale, M. E.), 653–659 (1973).
Ahmadjian, V. The Lichen Symbiosis (Wiley, 1993).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kang, D. D. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Seppey, M., Manni, M., & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. in Gene Prediction (ed. Kollmar, M.), 227–245 (2019).
Hiltunen, M., Ament-Velásquez, S. L. & Johannesson, H. The assembled and annotated genome of the fairy-ring fungus Marasmius oreades. Genome Biol. Evol. 13, 126 (2021).
Palmer, J. M., & Stajich, J. Funannotate v1. 8.1: Eukaryotic genome annotation. https://doi.org/10.5281/zenodo4054262 (2020).
Frith, M. C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 39, e23 (2011).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225 (2003).
Testa, A. C., Hane, J. K., Ellwood, S. R. & Oliver, R. P. CodingQuarry: Highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics 16, 1–12 (2015).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 1–9 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, 1–22 (2008).
Chan, P. P., & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. in Gene Prediction (ed. Kollmar, M.) 1–14 (2019).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D532 (2023).
Rawlings, N. D., Waller, M., Barrett, A. J. & Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 42, D503–D509 (2014).
Yin, Y. et al. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451 (2012).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Res. 51, D418–D427 (2023).
Blin, K. et al. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35 (2021).
O’Neill, E. Mining natural product biosynthesis in eukaryotic algae. Mar. Drugs 18, 90 (2020).
Lang, B. F. et al. Mitochondrial genome annotation with MFannot: A critical analysis of gene identification and gene model prediction. Front. Plant Sci. 14, 1222186 (2023).
Tillich, M. et al. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11 (2017).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Tagirdzhanova, G. et al. Evidence for a core set of microbial lichen symbionts from a global survey of metagenomes. Biorxiv https://doi.org/10.1101/2023.02.02.524463v1 (2023).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Yoshimura, I., Yamamoto, Y., Nakano, T., & Finnie, J. Isolation and culture of lichen photobionts and mycobionts. in Protocols in Lichenology: Culturing, Biochemistry, Ecophysiology and Use in Biomonitoring (eds. Kranner, I., Beckett, R. P., Varma, A.) 3–33 (2002).
U’Ren, J. M., Lutzoni, F., Miadlikowska, J. & Arnold, A. E. Community analysis reveals close affinities between endophytic and endolichenic fungi in mosses and lichens. Microb. Ecol. 60, 340–353 (2010).
Díaz-Escandón, D. et al. Genome-level analyses resolve an ancient lineage of symbiotic ascomycetes. Curr. Biol. 32, 5209–5218 (2022).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit–interactive quality assessment of genome assemblies. Genes Genomes Genet. 10, 1361–1374 (2020).
Acknowledgements
This work was supported by grants from the Leverhulme Trust RPG-2018-139, The Gatsby Charitable Foundation, the Halpin Family and the Biotechnology and Biological Sciences Research Council BBS/E/J/000PR9798 to N.J.T. The authors thank Paul Dyer and Peter Crittenden for providing the algal culture. The authors thank Camille Puginier, Francesco Dal Grande, and other authors of Puginier et al. [29] for sharing with us genome statistics behind Figs. 1 and 2 of their study. We thank Pavel Škaloud for discussions, and Phil Robinson and Jason Hollinger for providing photographs.
Author information
Authors and Affiliations
Contributions
G.T. and N.J.T. designed the study. G.T., K.S., and X.Y. performed experimental work. G.T. performed bioinformatic analysis. X.Y. and G.T. performed microscopy. G.T. and N.J.T. drafted the manuscript, and all authors contributed to editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tagirdzhanova, G., Scharnagl, K., Yan, X. et al. Genomic analysis of Coccomyxa viridis, a common low-abundance alga associated with lichen symbioses. Sci Rep 13, 21285 (2023). https://doi.org/10.1038/s41598-023-48637-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48637-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
