
Abstract
Fusarium species are among the most important phytopathogenic and toxigenic fungi. To understand the molecular underpinnings of pathogenicity in the genus Fusarium, we compared the genomes of three phenotypically diverse species: Fusarium graminearum, Fusarium verticillioides and Fusarium oxysporum f. sp. lycopersici. Our analysis revealed lineage-specific (LS) genomic regions in F. oxysporum that include four entire chromosomes and account for more than one-quarter of the genome. LS regions are rich in transposons and genes with distinct evolutionary profiles but related to pathogenicity, indicative of horizontal acquisition. Experimentally, we demonstrate the transfer of two LS chromosomes between strains of F. oxysporum, converting a non-pathogenic strain into a pathogen. Transfer of LS chromosomes between otherwise genetically isolated strains explains the polyphyletic origin of host specificity and the emergence of new pathogenic lineages in F. oxysporum. These findings put the evolution of fungal pathogenicity into a new perspective.
Similar content being viewed by others

Main
Fusarium species are among the most diverse and widely dispersed plant-pathogenic fungi, causing economically important blights, root rots or wilts1. Some species, such as F. graminearum (Fg) and F. verticillioides (Fv), have a narrow host range, infecting predominantly the cereals (Fig. 1a). By contrast, F. oxysporum (Fo), has a remarkably broad host range, infecting both monocotyledonous and dicotyledonous plants2 and is an emerging pathogen of immunocompromised humans3 and other mammals4. Aside from their differences in host adaptation and specificity, Fusarium species also vary in reproductive strategy. Some, such as Fo, are asexual, whereas others are both asexual and sexual with either self-fertility (homothallism) or obligate out-crossing (heterothallism) (Fig. 1b).

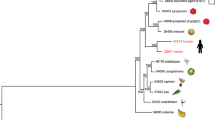
a, Maximum-likelihood tree using concatenated protein sequences of 100 genes randomly selected from 4,694 Fusarium orthologous genes that have clear 1:1:1:1 correlation among the Fusarium genomes and have unique matches in Magnaporthe grisea, Neurospora crassa and Aspergillus nidulans. The tree was constructed with PHYML35 (WAG model of evolution36). Branches are labelled with the percentage of 10,000 bootstrap replicates. b–d, Phenotypic variation within the genus Fusarium: b, disease symptoms of (top to bottom) kernel rot of maize (Fv), wilt of tomato (Fol), head blight of wheat (Fg) and root rot of pea (Fs); c, the perithecial states of Fv (Gibberella moniliformis), Fol (no sexual state), Fg (G. zeae) and Fs (Nectria haematococca); and d, micro- and macroconidia of Fv, Fol, Fg and Fs. Scale bars, 10 µm. Fg produces only macroconidia.
Previously, the genome of the cereal pathogen Fg was sequenced and shown to encode a larger number of proteins in pathogenicity related protein families compared to non-pathogenic fungi, including predicted transcription factors, hydrolytic enzymes, and transmembrane transporters5. We sequenced two additional Fusarium species, Fv, a maize pathogen that produces fumonisin mycotoxins that can contaminate grain, and F. oxysporum f.sp. lycopersici (Fol), a tomato pathogen. Here we present the comparative analysis of the genomes of these three species.
Results
Genome organization and gene clusters
We sequenced Fv strain 7600 and Fol strain 4287 (Methods, Supplementary Table 1) using a whole-genome shotgun approach and assembled the sequence using Arachne (Table 1, ref. 6). Chromosome level ordering of the scaffolds was achieved by anchoring the assemblies either to a genetic map for Fv (ref. 7), or an optical map for Fol (Supplementary Information A and Supplementary Table 2). We predicted Fol and Fv genes and re-annotated a new assembly of the Fg genome using a combination of manual and automated annotation (Supplementary Information B). The Fol genome (60 megabases) is about 44% larger than that of its most closely related species, Fv (42 Mb), and 65% larger than that of Fg (36 Mb), resulting in a greater number of protein-encoding genes in Fol (Table 1).
The relatedness of the three Fusarium genomes enabled the generation of large-scale unambiguous alignments (Supplementary Figs 1–3) and the determination of orthologous gene sets with high confidence (Methods, Supplementary Information C). On average, Fol and Fv orthologues display 91% nucleotide sequence identity, and both have 85% identity with Fg counterparts (Supplementary Fig. 4). Over 9,000 conserved syntenic orthologues were identified among the three genomes. Compared to other ascomycete genomes, these three-species orthologues are enriched for predicted transcription factors (P = 2.6 × 10-6), lytic enzymes (P = 0.001), and transmembrane transporters (P = 7 × 10-9) (Supplementary Information C and Supplementary Tables 3–8), in agreement with results reported for the Fg genome5.
Fusarium species produce diverse secondary metabolites, including mycotoxins that exhibit toxicity to humans and other mammals8. In the three genomes, we identified a total of 46 secondary metabolite biosynthesis (SMB) gene clusters. Microarray analyses confirmed the co-expression of genes in 14 of 18 Fg and 10 of 16 Fv SMB gene clusters. Ten out of the 14 Fg and eight out of the 10 Fv co-expressed SMB gene clusters are novel (Supplementary Information D, Supplementary Fig. 5 and Supplementary Table 9, and online materials), emphasizing the potential impact of uncharacterized secondary metabolites on fungal biology.
Lineage-specific chromosomes and pathogenicity
The genome assembly of Fol has 15 chromosomes, the Fv assembly 11 and the Fg assembly only four (Table 1). The smaller number of chromosomes in Fg is the result of chromosome fusion relative to Fv and Fo, and fusion sites in Fg match previously described high diversity regions (Supplementary Fig. 3, ref. 5). Global comparison among the three Fusarium genomes shows that the increased genomic territory in Fol is due to additional, unique sequences that reside mostly in extra chromosomes. Syntenic regions in Fol cover approximately 80% of the Fg and more than 90% of the Fv genome (Supplementary Information E and Supplementary Table 10), referred to as the ‘core’ of the genomes. Except for telomere-proximal regions, all 11 mapped chromosomes in the Fv assembly (41.1 Mb) correspond to 11 of the 15 chromosomes in Fol (41.8 Mb). The co-linear order of genes between Fol and Fv has been maintained within these chromosomes, except for one chromosomal translocation event and a few local rearrangements (Fig. 2a).

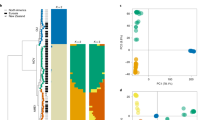
a, Argo37 dotplot of pair-wise MEGABLAST alignment (1 × 10-10) between Fv and Fol showing chromosome correspondences between the two genomes in the black dashed boxes. The vertical blue lines illustrate the chromosomal translocations, and the red dashed horizontal boxes highlight the Fol LS chromosomes. b, Global view of syntenic alignments between Fol and Fv and the distribution of transposable elements. Fol linkage groups are shown as the reference, and the length of the light grey background for each linkage group is defined by the Fol optical map. For each chromosome, row i represents the genomic scaffolds positioned on the optical linkage groups separated by scaffold breaks. Scaffold numbers for Fol are given above the blocks; row ii displays the syntenic mapping of Fv chromosomes, with one major translocation between chr 4/chr 12 in Fol and chr 4/chr 8 in Fv; row iii represents the density of transposable elements calculated with a 10 kb window. LS chromosomes include four entire chromosomes (chr 3, chr 6, chr 14 and chr 15) and parts of chromosome 1 and 2 (scaffold 27, scaffold 31), which lack similarity to syntenic chromosomes in Fv but are enriched for TEs. c, Two of the four Fol LS chromosomes showing the inter- (green) and intra- (yellow) chromosomal segmental duplications. The three traces below are density distribution of TEs (blue lines), secreted protein genes (green lines) and lipid metabolism related genes (red line). Chr, chromosome; Un, unmapped.
The unique sequences of Fol are a substantial fraction (40%) of the Fol assembly, designated as Fol lineage-specific (Fol LS) regions, to distinguish them from the conserved core genome. The Fol LS regions include four entire chromosomes (chromosomes 3, 6, 14 and 15), parts of chromosome 1 and 2 (scaffold 27 and scaffold 31, respectively), and most of the small scaffolds not anchored to the optical map (Fig. 2b). In total, the Fol LS regions encompass 19 Mb, accounting for nearly all of the larger genome size of Fol.
Notably, the LS regions contain more than 74% of the identifiable transposable elements (TEs) in the Fol genome, including 95% of all DNA transposons (Fig. 2b, Supplementary Fig. 6 and Supplementary Table 11). In contrast to the low content of repetitive sequence and minimal amount of TEs in the Fv and Fg genomes (Table 1 and Supplementary Table 11), about 28% of the entire Fol genome was identified as repetitive sequence (Methods), including many retroelements (copia-like and gypsy-like LTR retrotransposons, LINEs (long interspersed nuclear elements) and SINEs (short interspersed nuclear elements) and DNA transposons (Tc1-mariner, hAT-like, Mutator-like, and MITEs) (Supplementary Information E.3), as well as several large segmental duplications. Many of the TEs are full-length and present as highly similar copies. Particularly well represented DNA transposon classes in Fol are pogo, hAT-like elements and MITEs (in total approximately 550, 200 and 350 copies, respectively). In addition, there are one intra-chromosomal and two inter-chromosomal segmental duplications, totalling approximately 7 Mb and resulting in three- or even fourfold duplications of some regions (Fig. 2c). Overall, these regions share 99% sequence identity (Supplementary Fig. 7), indicating recent duplication events.
Only 20% of the predicted genes in the Fol LS regions could be functionally classified on the basis of homology to known proteins. These genes are significantly enriched (P < 0.0001) for the functional categories ‘secreted effectors and virulence factors’, ‘transcription factors’, and ‘proteins involved in signal transduction’, but are deficient in genes for house-keeping functions (Supplementary Information E and Supplementary Tables 12–18). Among the genes with a predicted function related to pathogenicity were known effector proteins (see below) as well as necrosis and ethylene-inducing peptides9 and a variety of secreted enzymes predicted to degrade or modify plant or fungal cell walls (Supplementary information E and Supplementary Tables 14, 15). Notably, many of these enzymes are expressed during early stages of tomato root infection (Supplementary Tables 15, 16 and Supplementary Fig. 8). The expansion of genes for lipid metabolism and lipid-derived secondary messengers in Fol LS regions indicates an important role for lipid signalling in fungal pathogenicity (Supplementary Fig. 9 and Supplementary Tables 13, 17). A family of transcription factor sequences related to FTF1, a gene transcribed specifically during early stages of infection of F. oxysporum f. sp. phaseoli (Supplementary Information E and Supplementary Table 4; ref. 10) is also expanded.
The recently published genome of F. solani11, a more diverged species, enabled us to extend comparative analysis to a larger evolutionary framework (Fig. 1). Whereas the ‘core’ genomes are well conserved among all four sequenced Fusarium species, the Fol LS regions are also absent in Fs (Supplementary Fig. 2). Additionally, Fs has three LS chromosomes distinct from the genome core11 and the Fol LS regions. In conclusion, each of the four Fusarium species carries a core genome with a high level of synteny whereas Fol and Fs each have LS chromosomes that are distinct with regard to repetitive sequences and genes related to host–pathogen interactions.
Origin of LS regions
Three possible explanations for the origin of LS regions in the Fol genome were considered: (1) Fol LS regions were present in the last common ancestor of the four Fusarium species but were then selectively and independently lost in Fv, Fg and Fs lineages during vertical transmission; (2) LS regions arose from the core genome by duplication and divergence within the Fol lineage; and (3) LS regions were acquired by horizontal transfer. To distinguish among these hypotheses, we compared the sequence characteristics of the genes in the Fol LS regions to those of genes in Fusarium core regions and genes in other filamentous fungi. If Fol LS genes have clear orthologues in the other Fusarium species, or paralogues in the core region of Fol, this would favour the vertical transmission or duplication with divergence hypotheses, respectively. We found that, whereas 90% of the Fol genes in the core regions have homologues in the other two Fusarium genomes, about 50% of the genes on Fol LS regions lack homologues in either Fv or Fg (1 × 10-20). Furthermore, there is less sequence divergence between Fol and Fv orthologues in core regions compared to Fol and Fg orthologues (Fig. 3a), consistent with the species phylogeny. In contrast, the LS genes that have homologues in the other Fusarium species are roughly equally distant from both Fv and Fg genes (Fig. 3b), indicating that the phylogenetic history of the LS genes differs from genes in the core region of the genome.

The scatter plots of BLAST score ratio (BSR)30 based on three-way comparisons of proteins encoded in core regions (a) and the Fol LS chromosomes (b). The numbers indicate the percentage of genes that lack homologous sequences in Fv and Fg (lower left corner), present in Fv but not Fg (x-axis) and present in Fg but not in Fv (y-axis). c, Discordant phylogenetic relationship of proteins encoded in the LS regions. The maximum-likelihood tree was constructed using the concatenated protein sequences of 100 genes randomly selected from 362 genes that share homologues in seven selected ascomycetes genomes including the four Fusarium genomes, M. grisea, N. crassa and A. nidulans. The trees were constructed with PHYML35 (WAG model of evolution36). The percentages for the branches represent the value based on a 10,000 bootstrapping data set.
Both codon usage tables and codon adaptation index (CAI) analysis indicate that the LS-encoding genes exhibit distinct codon usage (Supplementary Information E.5, Supplementary Fig. 10 and Supplementary Table 19) compared to the conserved genes and the genes in the Fv genome, further supporting their distinct evolutionary origins. The most significant differences were observed for amino acids Gln, Cys, Ala, Gly, Val, Glu and Thr, with a preference for G and C over A and T among the Fol LS genes (Supplementary Table 20). Such GC bias is also reflected in the slightly higher GC-content in their third codon positions (Supplementary Fig. 11).
Of the 1,285 LS-encoded proteins that have homologues in the NCBI protein set, nearly all (93%) have their best BLAST hit to other ascomycete fungi (Supplementary Fig. 12), indicating that Fol LS regions are of fungal origin. Phylogenetic analysis based on concatenated sampling of the 362 proteins that share homologues in seven selected ascomycete genomes — including the four sequenced Fusarium genomes, Magnaporthe grisea12, Neurospora crassa13 and Aspergillus nidulans14 — places their origin within the genus Fusarium but basal to the three most closely related Fusarium species Fg, Fv and Fol (Fig. 3c, Supplementary Table 21). Taken together, we conclude that horizontal acquisition from another Fusarium species is the most parsimonious explanation for the origin of Fol LS regions.
LS regions and host specificity
F. oxysporum is considered a species complex, composed of many different asexual lineages that can be pathogenic towards different hosts or non-pathogenic. The Fol LS regions differ considerably in sequence among Fo strains with different host specificities, as determined by Illumina sequencing of Fo strain Fo5176, a pathogen of Arabidopsis15 and EST (expressed sequence tag) sequences from Fo f. sp. vasinfectum16, a pathogen of cotton (Supplementary Information E.2). Despite less than 2% overall sequence divergence between shared sequences of Fol and Fo5176 (Supplementary Fig. 13A), for most of the sequences in the Fol LS regions there is no counterpart in Fo5176. (Supplementary Fig. 13B). Also Fov EST sequences16 have very high nucleotide sequence identity to the Fol genome (average 99%), but only match the core regions of Fol (Supplementary Information E.2). Large-scale genome polymorphism within Fo is also evident by differences in karyotype between strains (Supplementary Fig. 14)17. Previously, small, polymorphic and conditionally dispensable chromosomes conferring host-specific virulence have been reported in the fungi Nectria haematococca18 and Alternaria alternata19. Small (<2.3 Mb) and variable chromosomes are absent in non-pathogenic F. oxysporum isolates (Supplementary Fig. 14), indicating that Fol LS chromosomes may also be specifically involved in pathogenic adaptation.
Transfer of Fo pathogenicity chromosomes
It is well documented that small proteins are secreted during Fol colonizing the tomato xylem system20,21 and at least two of these, Six1 (Avr3) and Six3 (Avr2), are involved in virulence functions22,23. Interestingly, the genes for these proteins, as well as a gene for an in planta-secreted oxidoreductase (ORX1)20, are located on chromosome 14, one of the Fol LS chromosomes. These genes are all conserved in strains causing tomato wilt, but are generally not present in other strains24. The genome data enabled the identification of the genes for three additional small in planta-secreted proteins on chromosome 14, named SIX5, SIX6 and SIX7 (Supplementary Table 22) based on mass spectrometry data obtained previously20. Together these seven genes can be used as markers to identify each of the three supercontigs (SC 22, 36 and 51) localized to chromosome 14 (Supplementary Table 23 and Supplementary Fig. 15).
In view of the combined experimental findings and computational evidence, we proposed that LS chromosome 14 could be responsible for pathogenicity of Fol towards tomato, and that its mobility between strains could explain its presence in tomato wilt pathogens, comprising several clonal lineages polyphyletic within the Fo species complex, but absence in other lineages24. To test these hypotheses, we investigated whether chromosome 14 could be transferred and whether the transfer would shift pathogenicity between different strains of Fo, using the genes for in planta-secreted proteins on chromosome 14 as markers. Fol007, a strain that is able to cause tomato wilt, was co-incubated with a non-pathogenic isolate (Fo-47) and two other strains that are pathogenic towards melon (Fom) or banana (Foc), respectively. A gene conferring resistance against zeocin (BLE) was inserted close to SIX1 as a marker to select for transfer of chromosome 14 from the donor strain into Fo-47, Fom or Foc. The receiving strains were transformed with a hygromycin resistance gene (HYG), inserted randomly into the genome; three independent hygromycin resistant transformants per recipient strain were selected. Microconidia of the different strains were isolated and mixed in a 1:1 ratio on agar plates. Spores emerging on these plates after 6–8 days of incubation were selected for resistance to both zeocin and hygromycin. Double drug-resistant colonies were recovered with Fom and Fo-47, but not using Foc as the recipient, at a frequency of roughly 0.1 to 10 per million spores (Supplementary Table 24).
Pathogenicity assays demonstrated that double drug-resistant strains derived from co-incubating Fol007 with Fo-47, referred to as Fo-47+, had gained the ability to infect tomato to various degrees (Fig. 4a, b). In contrast, none of the double drug-resistant strains derived from co-incubating Fol007 with Fom were able to infect tomato. All Fo-47+ strains contained large portions of Fol chromosome 14 as demonstrated by PCR amplification of the seven gene markers (Fig. 4c, Supplementary Fig. 15 and Supplementary Information F). The parental strains, as well as the sequenced strain Fol4287, each have distinct karyotypes. This enabled us to determine with chromosome electrophoresis whether the entire chromosome 14 of Fol007 was transferred into Fo-47+ strains. All Fo-47+ strains had the same karyotype as Fo-47, except for the presence of one or two additional small chromosomes (Fig. 4d). The chromosome present in all Fo-47+ strains (Fig. 4d, arrow number 1) was confirmed to be chromosome 14 from Fol007 based on its size and a Southern hybridization using a SIX6 probe (Fig. 4e). Interestingly, two double drug-resistant strains (Fo-47+ 1C and Fo-47+ 2A in Fig. 4a), which caused the highest level of disease (Fig. 4a, b), have a second extra chromosome, corresponding in size to the smallest chromosome in the donor strain Fol007 (Fig. 4d, arrow number 2).

a, Tomato plants infected with Fol007, Fo-47 or double drug resistant Fo-47+ strains (1A through 3C) derived from this parental combination, two weeks after inoculation as described for b. b, Eight of nine Fo-47+ strains derived from pairing Fol007 and Fo-47 show pathogenicity towards tomato. Average disease severity in tomato seedlings was measured 3 weeks after inoculation in arbitrary units (a.u.). The overall phenotype and the extent of browning of vessels was scored on a scale of 0–4: 0, no symptoms; 1, slightly swollen and/or bent hypocotyl; 2, one or two brown vascular bundles in hypocotyl; 3, at least two brown vascular bundles and growth distortion (strong bending of the stem and asymmetric development); 4, all vascular bundles are brown, plant either dead or stunted and wilted. c, The presence of SIX genes and ORX1 in Fom, Fo-47 and Fol isolates and in double drug-resistant strains derived from co-incubation of Fol/Fom and Fol/Fo-47, assessed by PCR on genomic DNA. Co-incubations were performed with the isolates shown in bold. Three independent transformants of Fom and Fo-47 with a randomly inserted hygromycin resistance gene (H1, H2, H3) were investigated. d, Fo-47+ strains derived from a Fol007/Fo-47 co-incubation have the same karyotype as Fo-47, plus one or two chromosomes from Fol007. Protoplasts from Fol4287, Fol007 (with BLE on chromosome 14), three independent HYG transformants of Fo-47 (lane Fo-47 H1, H2 and H3) and nine Fo-47+strains (lane 1A to 3C, the number 1, 2 or 3 referring to the HYG resistant transformant from which they were derived) were loaded on a CHEF (contour-clamped homogeneous electric field) gel. Chromosomes of S. pombe were used as a molecular size marker. Arrows 1 and 2 point to additional chromosomes in the Fo-47+ strains relative to Fo-47. e, Southern blot of the CHEF gel shown in d, hybridized with a SIX6 probe, showing that chromosome 14 (arrow 1 in d) is present in all strains except Fo-47 (H1, H2 and H3).
To rigorously assess whether additional genetic material other than chromosome 14 may have been transferred from Fol007 into Fo-47+ strains, we developed PCR primers for amplification of 29 chromosome-specific markers from Fol007 but not Fo-47. These markers (on average two for each chromosome) were used to screen Fo-47+ strains for the presence of Fol007-derived genomic regions (Supplementary information F.4 and Supplementary Fig. 16). All Fo-47+ strains were shown to have the chromosome 14 markers (Supplementary Fig. 17), but not Fol007 markers located on any core chromosome, confirming that core chromosomes were not transferred. Interestingly, the two Fo-47+ strains (1C and 2A) that have the second small chromosome and caused more disease symptoms were also positive for an additional Fol007 marker (Supplementary Fig. 17), associated with a large duplicated LS region in Fol4287: scaffold 18 (1.3 Mb on chromosome 3) and scaffold 21 (1.0 Mb on chromosome 6) (Fig. 2c). The presence of most or all of the sequence of scaffold 18/21 in strains 1C and 2A was confirmed with an additional nine primer pairs for genetic markers scattered over this region (data not shown, see Supplementary Tables 25a, b for primer sequences) (Fig. 4d).
Taken together, we conclude that pathogenicity of Fo-47+ strains towards tomato can be specifically attributed to the acquisition of Fol chromosome 14, which contains all known genes for small in planta-secreted proteins. In addition, genes on other LS chromosomes may further enhance virulence as demonstrated by the two strains containing the additional LS chromosome from Fol007. We did not find a double drug-resistant strain with a tagged chromosome of Fo-47 in the Fol007 background. Also, a randomly tagged transformant of Fol007 did not render any double drug-resistant colonies when co-incubated with Fo-47 (data not shown). This indicates that transfer between strains may be restricted to certain chromosomes, perhaps determined by various factors, including size and TE content of the chromosome. Their propensity for transfer is supported by the fact that the smallest LS chromosome in Fol007 moved to Fo-47 without being selected for drug resistance in two out of nine cases.
Discussion
Comparison of Fusarium genomes revealed a remarkable genome organization and dynamics of the asexual species Fol. This tomato pathogen contains four unique chromosomes making up more than one-quarter of its genome. Sequence characteristics of the genes in the LS regions indicate a distinct evolutionary origin of these regions. Experimentally, we have demonstrated the transfer of entire LS chromosomes through simple co-incubation between two otherwise genetically isolated members of Fo. The relative ease by which new tomato pathogenic genotypes are generated supports the hypothesis that such transfer between Fo strains may have occurred in nature24 and has a direct impact on our understanding of the evolving nature of fungal pathogens. Although rare, horizontal gene transfer has been documented in other eukaryotes, including metazoans25. However, spontaneous horizontal transfer of such a large portion of a genome and the direct demonstration of associated transfer of host-specific pathogenicity has not been previously reported.
Horizontal transfer of host specificity factors between otherwise distant and genetically isolated lineages of Fo may explain the apparent polyphyletic origins of host specialization26 and the rapid emergence of new pathogenic lineages in otherwise distinct and incompatible genetic backgrounds27. Fol LS regions are enriched for genes related to host–pathogen interactions. The mobilization of these chromosomes could, in a single event, transfer an entire suite of genes required for host compatibility to a new genetic lineage. If the recipient lineage had an environmental adaptation different from the donor, transfer could increase the overall incidence of disease in the host by introducing pathogenicity in a genetic background pre-adapted to a local environment. Such knowledge of the mechanisms underpinning rapid pathogen adaptation will affect the development of strategies for disease management in agricultural settings.
Methods Summary
Generation of genome sequencing and assembly
The whole genome shotgun (WGS) assemblies of Fv (8× coverage) and Fol (6.8× coverage) were generated using Sanger sequencing technology and assembled using Arachne6. Physical maps were created by anchoring the assemblies to the Fv genetic linkage map7 and to the Fol optical map, respectively.
Defining hierarchical synteny
Local-alignment anchors were detected using PatternHunter (1 × 1010) (ref. 28). Contiguous sets of anchors with conserved order and orientation were chained together within 10 kb distance and filtered to ensure that no block overlaps another block by more than 90% of its length.
Identification of repetitive sequences
Repeats were detected by searching the genome sequence against itself using CrossMatch (≥ 200 bp and ≥ 60% sequence similarity). Full-length TEs were annotated using a combination of computational predictions and manual inspection. Large segmental duplications were identified using Map Aligner29.
Characterization of proteomes
Orthologous genes were determined based on BLASTP and pair-wise syntenic alignments (SI). The blast score ratio tests30 were used to compare relatedness of proteins among three genomes. The EMBOSS tool ‘cusp’ (http://emboss.sourceforge.net/) was used to calculate codon usage frequencies. Gene Ontology terms were assigned using Blast2GO31 software (BLASTP 1 × 1020) and tested for enrichment using Fisher’s exact test, corrected for multiple testing32. A combination of homology search and manual inspection was used to characterize gene families33,34. Potentially secreted proteins were identified using SignalP (http://www.cbs.dtu.dk/services/SignalP/) after removing trans-membrane/mitochondrial proteins based with TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), Phobius (except in the first 50 amino acids), and TargetP (RC score 1 or 2) predictions. Small cysteine-rich secreted proteins were defined as secreted proteins that are less than 200 amino acids in length and contain at least 4% cysteine residues. GPI (glycosyl phosphatidyl inositol)-anchor proteins were identified by the GPI-anchor attachment signal among the predicted secreted proteins using a custom PERL script.
Accession codes
Data deposits
All sequence reads can be downloaded from the NCBI trace repository. The assemblies of Fv and Fol have been deposited at GenBank under the project accessions AAIM02000000 and AAXH01000000. Detailed information can be accessed through the Broad Fusarium comparative website: http://www.broad.mit.edu/annotation/genome/fusarium_group.3/MultiHome.html.
References
Agrios, G. N. Plant Pathology 5th edn (Academic Press, 2005)
Armstrong, G. M. & Armstrong, J. K. in Fusarium: Diseases, Biology and Taxonomy (eds Nelson, P. E., Toussoun, T. A. & Cook R.) 391–399 (Penn State University Press, 1981)
O’Donnell, K. et al. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J. Clin. Microbiol. 42, 5109–5120 (2004)
Ortoneda, M. et al. Fusarium oxysporum as a multihost model for the genetic dissection of fungal virulence in plants and mammals. Infect. Immun. 72, 1760–1766 (2004)
Cuomo, C. A. et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317, 1400–1402 (2007)
Jaffe, D. B. et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13, 91–96 (2003)
Xu, J. R. & Leslie, J. F. A genetic map of Gibberella fujikuroi mating population A (Fusarium moniliforme). Genetics 143, 175–189 (1996)
Desjardins, A. E. & Proctor, R. H. Molecular biology of Fusarium mycotoxins. Int. J. Food Microbiol. 119, 47–50 (2007)
Qutob, D. et al. Phytotoxicity and innate immune responses induced by Nep1-like proteins. Plant Cell 18, 3721–3744 (2006)
Ramos, B. et al. The gene coding for a new transcription factor (ftf1) of Fusarium oxysporum is only expressed during infection of common bean. Fungal Genet. Biol. 44, 864–876 (2007)
Coleman, J. J. et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 5, e1000618 (2009)
Dean, R. A. et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434, 980–986 (2005)
Galagan, J. E. et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422, 859–868 (2003)
Galagan, J. E. et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438, 1105–1115 (2005)
Thatcher, L. F., Manners, J. M. & Kazan, K. Fusarium oxysporum hijacks COI1-mediated jasmonate signaling to promote disease development in Arabidopsis. Plant J. 58, 927–939 (2009)
Dowd, C., Wilson, I. W. & McFadden, H. Gene expression profile changes in cotton root and hypocotyl tissues in response to infection with Fusarium oxysporum f. sp. vasinfectum. Mol. Plant Microbe Interact. 17, 654–667 (2004)
Teunissen, H. A. et al. Construction of a mitotic linkage map of Fusarium oxysporum based on Foxy-AFLPs. Mol. Genet. Genomics 269, 215–226 (2003)
Miao, V. P., Covert, S. F. & VanEtten, H. D. A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science 254, 1773–1776 (1991)
Harimoto, Y. et al. Expression profiles of genes encoded by the supernumerary chromosome controlling AM-toxin biosynthesis and pathogenicity in the apple pathotype of Alternaria alternata. Mol. Plant Microbe Interact. 20, 1463–1476 (2007)
Houterman, P. M. et al. The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants. Mol. Plant Pathol. 8, 215–221 (2007)
van der Does, H. C. et al. Expression of effector gene SIX1 of Fusarium oxysporum requires living plant cells. Fungal Genet. Biol. 45, 1257–1264 (2008)
Houterman, P. M. et al. The effector protein Avr2 of the xylem colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J. 58, 970–978 (2009)
Rep, M. et al. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 53, 1373–1383 (2004)
van der Does, H. C. et al. The presence of a virulence locus discriminates Fusarium oxysporum isolates causing tomato wilt from other isolates. Environ. Microbiol. 10, 1475–1485 (2008)
Gladyshev, E. A., Meselson, M. & Arkhipova, I. R. Massive horizontal gene transfer in bdelloid rotifers. Science 320, 1210–1213 (2008)
O’Donnell, K., Kistler, H. C., Cigelnik, E. & Ploetz, R. C. Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl Acad. Sci. USA 95, 2044–2049 (1998)
Gale, L. R., Katan, T. & Kistler, H. C. The probable center of origin of Fusarium oxysporum f. sp. lycopersici VCG 0033. Plant Dis. 87, 1433–1438 (2003)
Li, M., Ma, B., Kisman, D. & Tromp, J. Patternhunter II: highly sensitive and fast homology search. J. Bioinform. Comput. Biol. 2, 417–439 (2004)
Zhou, S. et al. Single-molecule approach to bacterial genomic comparisons via optical mapping. J. Bacteriol. 186, 7773–7782 (2004)
Rasko, D. A., Myers, G. S. & Ravel, J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics 6, 2 (2005)
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674 (2005)
Blüthgen, N. et al. Biological profiling of gene groups utilizing Gene Ontology. Genome Inform 16, 106–115 (2005)
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37 (Database issue). D233–D238 (2009)
Miranda-Saavedra, D. & Barton, G. J. Classification and functional annotation of eukaryotic protein kinases. Proteins 68, 893–914 (2007)
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003)
Whelan, S. & Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 (2001)
Engels, R. et al. Combo: a whole genome comparative browser. Bioinformatics 22, 1782–1783 (2006)
Acknowledgements
The 4× sequence of F. verticillioides was provided by Syngenta Biotechnology Inc. Generation of the other 4× sequence of F. verticillioides and 6.8× sequence of F. oxysporum f. sp. lycopersici was funded by the National Research Initiative of USDA's National Institute of Food and Agriculture through the Microbial Genome Sequencing Program (2005-35600-16405) and conducted by the Broad Institute Sequencing Platform. Wayne Xu and the Minnesota Supercomputing Institute for Advanced Computational Research are also acknowledged for their support. The authors thank Leslie Gaffney at the Broad Institute for graphic design and editing and Tracy E. Anderson of the University of Minnesota, College of Biological Sciences Imaging Center for spore micrographs.
Author Contributions L.-J.M., H.C.D., M.R. and H.C.K. coordinated genome annotation, data analyses, experimental validation and manuscript preparation. L.-J.M. and H.C.D. made equivalent contributions and should be considered joint first authors. H.C.K. and M.R. contributed equally as corresponding authors. K.A.B., C.A.C., J.J.C., M.-J.D., A.D.P., M.D., M.F., J.G., M.G., B.H., P.M.H., S.K., W.-B.S., C.W., X.X. and J.-R.X. made major contributions to genome sequencing, assembly, analyses and production of complementary data and resources. All other authors are members of the genome sequencing consortium and contributed annotation, analyses or data throughout the project.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Information on (A) Genome sequencing, assembling and mapping; (B) Annotation; (C) Features common to Fusarium genomes; (D) Secondary metabolism gene clusters; (E) Features of Fusarium oxysporum LS chromosomes and (F) Transfer of LS chromosomes between strains. (PDF 522 kb)
Supplementary Figures
This file contains Supplementary Figures S1-S20 with legends. (PDF 2911 kb)
Supplementary Tables
This file contains Supplementary Tables S1-S25. (PDF 679 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Ma, LJ., van der Does, H., Borkovich, K. et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373 (2010). https://doi.org/10.1038/nature08850
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature08850
This article is cited by
-
Core hyphosphere microbiota of Fusarium oxysporum f. sp. niveum
Environmental Microbiome (2024)
-
Whole-genome sequencing of Fusarium spp. causing sugarcane root rot on both chewing cane and sugar-making cane
Stress Biology (2024)
-
Molecular characterization of Indian races of Fusarium oxysporum f. sp. lentis (Fol) based on secreted in Xylem (SIX) effector genes and development of a SIX11 gene-based molecular marker for specific detection of Fol
Archives of Microbiology (2024)
-
Potential application of a fungal co-culture crude extract for the conservation of post-harvest fruits
Brazilian Journal of Microbiology (2024)
-
Whole genome sequencing and analysis of Armillaria gallica Jzi34 symbiotic with Gastrodia elata
BMC Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
