
Abstract
Candida species are the most common cause of opportunistic fungal infection worldwide. Here we report the genome sequences of six Candida species and compare these and related pathogens and non-pathogens. There are significant expansions of cell wall, secreted and transporter gene families in pathogenic species, suggesting adaptations associated with virulence. Large genomic tracts are homozygous in three diploid species, possibly resulting from recent recombination events. Surprisingly, key components of the mating and meiosis pathways are missing from several species. These include major differences at the mating-type loci (MTL); Lodderomyces elongisporus lacks MTL, and components of the a1/α2 cell identity determinant were lost in other species, raising questions about how mating and cell types are controlled. Analysis of the CUG leucine-to-serine genetic-code change reveals that 99% of ancestral CUG codons were erased and new ones arose elsewhere. Lastly, we revise the Candida albicans gene catalogue, identifying many new genes.
Similar content being viewed by others


Main
Four Candida species, C. albicans, C. glabrata, C. tropicalis and C. parapsilosis, together account for ∼95% of identifiable Candida infections1. Although C. albicans is still the most common causative agent, its incidence is declining and the frequency of other species is increasing. Of these, C. parapsilosis is a particular problem in neonates, transplant recipients and patients receiving parenteral nutrition; C. tropicalis is more commonly associated with neutropenia and malignancy. Other Candida species, including C. krusei, C. lusitaniae and C. guilliermondii, account for <5% of invasive candidiasis. Almost all Candida species, with the exceptions of C. glabrata and C. krusei, belong in a single Candida clade (Fig. 1) characterized by the unique translation of CUG codons as serine rather than leucine2. Within this, haploid and diploid species occupy two separate subclades (Fig. 1).

Tree topology and branch lengths were inferred with MRBAYES (Supplementary Information, section S5). Posterior probabilities are indicated for each branch. The asterisk marks a branch that was constrained on the basis of syntenic conservation40.
To determine the genetic features underlying their diversity of biology and pathogenesis, we sequenced six genomes from the Candida clade (Fig. 1). These include a second sequenced isolate of C. albicans (WO-1) characterized for white–opaque switching, a phenotypic change that correlates with host specificity and mating3,4. We also sequenced the major pathogens C. tropicalis and C. parapsilosis; L. elongisporus, a close relative of C. parapsilosis recently identified as a cause of bloodstream infection5; and two haploid emerging pathogens, C. guilliermondii and C. lusitaniae. We compared these with the previously sequenced C. albicans strain (SC5314)6,7,8, Debaryomyces hansenii9, a marine yeast rarely associated with disease, and nine species from the related Saccharomyces clade (Fig. 1). These species span a wide evolutionary range and show large phenotypic differences in pathogenicity and mating, allowing us to study the genomic basis for these traits.
Genome sequence and comparative annotation
We found enormous variation in genome size and composition between the Candida genomes sequenced (Table 1). Each genome assembly displayed high continuity, ranging from nine to 27 scaffolds (Supplementary Table 1). Scaffold number and size largely match pulsed-field gel electrophoresis estimates for all genomes, and telomeric repeat arrays are linked to the ends of nearly all large scaffolds (Supplementary Information, section S2). Genome size ranges from 10.6 to 15.5 megabases (Mb), a difference of nearly 50%, with haploid species having smaller genomes. GC content ranges from 33% to 45% (Table 1). Transposable elements and other repetitive sequences vary in number and type between assemblies (Supplementary Information, section S6). Regions similar to the major repeat sequence (MRS) of C. albicans were found only in C. tropicalis, suggesting that MRS-associated recombination could contribute to the observed karyotypic variation among C. tropicalis strains10.
Despite the genome size and phenotypic variation among the species, the predicted numbers of protein-coding genes are very similar, ranging from 5,733 to 6,318 (Table 1). Even the small differences in gene number are not correlated with genome size; the smallest genome, C. guilliermondii, has more genes than the largest genome, L. elongisporus. Instead, genome size differences are explained by an approximately threefold variation in intergenic spacing (Table 1). Large syntenic blocks of conserved gene order were detected among the four diploid species and between two of the haploid species, C. guilliermondii and D. hansenii (Supplementary Fig. 5). Although synteny blocks have been shuffled by local inversions and rearrangements, these have been primarily intrachromosomal as chromosome boundaries have been largely preserved across the diploid genomes (Supplementary Fig. 5).
Given the high conservation of protein-coding genes across the Candida clade, we used multiple alignments of the related genomes to revise the annotation of C. albicans. We identified 91 new or updated genes, of which 80% are specific to the Candida clade (Supplementary Information, section S4). We also corrected existing annotations in C. albicans, revealing 222 dubious genes, and also identified 190 probable frame shifts and 36 nonsense sequencing errors in otherwise well-conserved genes (Supplementary Information, section S4). In each case, manual curation confirmed ∼80% of these predictions.
Polymorphism in diploid genomes
To gain insights into the recent history of C. albicans, we compared the two diploid strains, SC5314 and WO-1, which belong to different population subgroups11. Variation in the karyotype of these strains is primarily due to translocations at MRS sequences (ref. 12 and Supplementary Fig. 1). The two assemblies are largely co-linear with 12 inversions of 5–94 kilobases between them, except that in WO-1 some non-homologous chromosomes have recombined at the MRS (Supplementary Information, section S8). We found similar rates of single nucleotide polymorphisms (SNPs) within each strain (one SNP per 330–390 bases), and twice this rate between them, suggesting relatively recent divergence. Polymorphism rates in the other diploids range from one SNP per 222 bases in L. elongisporus and one SNP per 576 bases in C. tropicalis to a particularly low one SNP per 15,553 bases in C. parapsilosis, more than 70-fold lower than in the closely related L. elongisporus.
Notable regions of extended homozygosity are found in three of the four diploid genomes, which may reflect break-induced replication or recent passage through a parasexual or sexual cycle. Candida albicans, C. tropicalis and L. elongisporus each shows large chromosomal regions devoid of SNPs, extending up to ∼1.2 Mb (Fig. 2 and Supplementary Figs 6–8). In contrast, the few SNPs in C. parapsilosis are randomly distributed across the genome (Supplementary Fig. 9a). A total of 4.3 Mb (30%) of the WO-1 assembly is homozygous for SNPs, approximately twice that found in SC5314 (Supplementary Information, section 7, and refs 7, 13, 14). There is at least one homozygous region per chromosome, none of which spans the predicted centromeres and only one of which starts at a MRS (Fig. 2). Whereas nearly all homogeneous regions are present at diploid levels and are therefore homozygous, WO-1 has lost one copy of a >300-kilobase region on chromosome 3 comprising nearly 200 genes (Supplementary Fig. 10). The pressure to maintain this region as homozygous in both strains is apparently high, as it is diploid but homogenous in SC5314.

Red lines show SNPs per kilobase, normalized by coverage, within WO-1, and blue lines show SNPs per kilobase between WO-1 and SC5314. Although both copies of chromosome 5 have rearranged at the MRS (yellow box) in WO-1, we show this as a single chromosome to allow a haploid reference for polymorphism. Relative to SC5314, chromosomes 1, 4 and 6 have the opposite orientation (Supplementary Fig. 6). Chr, chromosome; sc, supercontig; kb, kilobase.
Usage and evolution of CUG codons
All Candida clade species translate CUG codons as serine instead of leucine15. This genetic-code change altered the decoding rules of CUN codons in the Candida clade: whereas Saccharomyces cerevisiae uses two transfer RNAs, each of which translates two codons, Candida species use a dedicated tRNACAGSer for CUG codons and a single tRNAIAGLeu for CUA, CUC, and CUU codons, as inosine can base-pair with A, C and U (Fig. 3). This alteration in decoding rules forced the reduced usage of CUG—and also CUA, probably as a result of the weaker wobble—in Candida genes (Fig. 3a). CUU and CUC codons do not display the same bias for infrequent usage (Fig. 3b). An additional pressure influencing codon usage may be the GC content, as usage of leucine codons in Candida species is correlated with the percentage GC composition (Supplementary Table 11).

a, Average percentage of genes with zero, one or two CUG and CUA codons in Candida (grey bars) and Saccharomyces (black bars). Error bars, s.d. All differences are significant with P ≤ 0.0004 (t-test, N = 8 Candida spp. and 6 Saccharomyces spp.) except for those in genes with two CUA codons (P = 0.02). ‘Candida’ and ‘Saccharomyces’ here refer to the CTG and WGD clades in Fig. 1, but including Pichia stipitis and excluding C. dubliniensis. b, CUN codon usage for all codon counts. c, Decoding rules for CUN codons in Saccharomyces and Candida.
We also examined the evolutionary fate of ancestral CUG codons and the origin of new CUG codons (Supplementary Table 12). CUG codons in C. albicans almost never (1%) align opposite CUG codons in S. cerevisiae. Instead, CUG serine codons in C. albicans align primarily to Saccharomyces codons for serine (20%) and other hydrophilic residues (49%). CUG leucine codons in S. cerevisiae align primarily to leucine codons in Candida (50%) and to other hydrophobic-residue codons (30%). This suggests a complete functional replacement of CUG codons in Candida.
Gene family evolution
To identify gene families likely to be associated with Candida pathogenicity and virulence, we used a phylogenomic approach across seven Candida and nine Saccharomyces genomes (Supplementary Information, section S10). Among 9,209 gene families, we identified 21 that are significantly enriched in the more common pathogens (Table 2). These include families encoding lipases, oligopeptide transporters and adhesins, which are all known to be associated with pathogenicity8, as well as poorly characterized families not previously associated with pathogenesis.
Three cell wall families are enriched in the pathogens: those encoding Hyr/Iff proteins (ref. 16), Als adhesins17 and Pga30-like proteins (Table 2 and Supplementary text S11). The Als family (family 17 in Supplementary Tables 22 and 23) in C. albicans is associated with virulence, and in particular with adhesion to host surfaces18, invasion of host cells19 and iron acquisition20. All these families are absent from the Saccharomyces clade species and are particularly enriched in the more pathogenic species (Supplementary Table 18). All three families are highly enriched for gene duplications (Supplementary Table 19), including tandem clusters of two to six genes, and show high mutation rates (fastest 5% of families) (Supplementary Information, section S10d). This variable repertoire of cell wall proteins is likely to be of profound importance to the niche adaptations and relative virulence of these organisms.
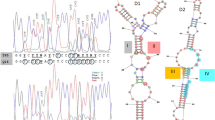
Als17 and Hyr/Iff genes frequently contain intragenic tandem repeats, which modulate adhesion and biofilm formation in S. cerevisiae21 (Fig. 4 and Supplementary Figs 19 and 20). The sequence of intragenic tandem repeats is conserved at the protein level across species (Supplementary Figs 19 and 20). Two proteins contain both an Als domain and repeats characteristic of the Hyr/Iff family.

The amino-terminal Als domain (red) and Hyr/Iff domain (green) are shown as ovals. Intragenic tandem repeats (ITRs; see Supplementary Information, section S11) are shown as rectangles, coloured to represent similar amino-acid sequences.
Candida clade pathogens show expansions of extracellular enzyme and transmembrane transporter families (Table 2 and Supplementary Table 22). These families are either not found in Saccharomyces (including amino-acid permeases, lipases and superoxide dismutases) or are present in S. cerevisiae but significantly expanded in pathogens (including phospholipase B, ferric reductases, sphingomyelin phosphodiesterases and GPI-anchored yapsin proteases, which have been linked to virulence in C. glabrata22). Several groups of cell-surface transporters are also enriched (including oligopeptide transporters, amino-acid permeases and the major facilitator superfamily). Overall, these family expansions illustrate the importance of extracellular activities in virulence and pathogenicity. Genes involved in stress response are also variable between species (Supplementary Information, section S12).
C. albicans also showed species-specific expansion of some families, including two associated with filamentous growth, a leucine-rich repeat family and the Fgr6-1 family (Table 2). Candida albicans forms hyphae whereas C. tropicalis and C. parapsilosis produce only pseudohyphae, so these families may contribute to differences in hyphal growth.
We identified 64 families showing positive selection in the highly pathogenic Candida species (Supplementary Table 32). These are highly enriched for cell wall, hyphal, pseudohyphal, filamentous growth and biofilm functions (Supplementary Information, section S13). Six of the families have been previously associated with pathogenesis, including that of ERG3, a C-5 sterol desaturase essential for ergosterol biosynthesis, for which mutations can cause drug resistance23.
Structure of the MTL locus
Pathogenic fungi may have limited their sexual cycles to maximize their virulence24, and the sequenced Candida species show tremendous diversity in their apparent abilities to mate. Among the four diploids, C. albicans has a parasexual cycle (mating of diploid cells followed by mitosis and chromosome loss instead of meiosis25), L. elongisporus has been described as sexual and homothallic (self-mating)26, whereas C. tropicalis and C. parapsilosis have never been observed to mate. Among the three haploids, C. guilliermondii and C. lusitaniae are heterothallic (cross-mating only) and have a complete sexual cycle, whereas D. hansenii is haploid and homothallic27 (Supplementary Information, section 14c).
To understand the genomic basis for this diversity, we studied the Candida MTL locus, which determines mating type, similar to the MAT locus in S. cerevisiae. In both C. albicans and S. cerevisiae, the mating locus has two idiomorphs, a and α, encoding the regulators a1, α1 and α2, respectively. Candida albicans MTL a also encodes a2, and both idiomorphs in this species contain alleles of three additional genes without known roles in mating: PAP, OBP and PIK28. The MTLα and MTL a genes, alone or in combination, specify one of the three possible cell-type programs (a haploid, α haploid, a/α diploid). In C. albicans, the alpha-domain protein α1 activates α-specific mating genes, the high-mobility group (HMG) factor a2 activates a-specific mating genes, and the a1/α2 homeodomain heterodimer represses mating genes in a/α cells29,30.
Despite extended conservation of the genomic context flanking the mating-type locus, there is great variability in MTL gene content (Fig. 5). MTL a 1 has become a pseudogene in C. parapsilosis31, and is probably a recent loss because target genes retain predicted a1/α2 binding sites (Supplementary Information, section S14). MTLα2 is missing in both C. guilliermondii and C. lusitaniae (J. L. Reedy, A. Floyd and J. Heitman, submitted). A fused mating-type locus containing both a and α genes is found in D. hansenii and Pichia stipitis32,33.

a, MTL a -specific genes are shown in grey, MTLα-specific genes are shown in black and other orthologues are shown in colour. Two idiomorphs from C. albicans and C. tropicalis are shown. Arrows indicate inversions relative to C. albicans. Crosses show gene losses and the C. parapsilosis MTL a 1 pseudogene. There is no MTL locus in L. elongisporus; gene order around the PAP a , OBP a and PIK a genes is shown. For C. guilliermondii and C. lusitaniae, the genome project sequenced one idiomorph; the second was obtained by J. Reedy. In D. hansenii, PAP, OBP and PIK are separate from the fused MTL locus32. b, Placement of gene losses on the phylogenetic tree. HOM, homothallic; HET, heterothallic.
Most surprisingly, all four mating-type genes are missing in L. elongisporus. It contains a site syntenic to MTL a in other species, but this contains only 508 base pairs of apparently non-coding DNA, a length insufficient to encode a1 or a2 even if they were extensively divergent in sequence. We confirmed this finding in seven other L. elongisporus isolates (not shown). The sexual state of L. elongisporus has been assumed to be homothallic because asci are generated from identical cells5,26, but the absence of an a-factor pheromone, receptor and transporter (Supplementary Table 34) as well as MTL, suggests that it may not have a sexual cycle. Alternatively, mating may require only one pheromone and receptor (α), and L. elongisporus may be the first identified ascomycete that can mate independently of MTL or MAT.
Our discovery that MTLα2 and MTL a 1 are frequently absent challenges our understanding of how the mating locus operates. The model derived for mating regulation in C. albicans, including the role of a1/α2 in white–opaque switching3, must differ substantially in the other sexual species. This is particularly interesting because the major regulators of the white–opaque switch in C. albicans (WOR1, WOR2, CZF1 and EFG134) are generally conserved in other species, but Wor1 control over white–opaque genes appears to be a recent innovation in C. albicans and Candida dubliniensis35. Our data also suggest that the loss of α2 occurred early in the haploid sexual lineage.
Mating and meiosis
To gain further insight into their diversity in sexual behaviour, we examined whether 227 genes required for meiosis in S. cerevisiae and other fungi have orthologues in the Candida species (Supplementary Information, section S14). A previous report36 that some components of meiosis, such as the major regulator IME1, are missing from C. albicans led us to propose that their loss could be correlated with lack of meiosis. Surprisingly, however, we find that these genes are missing in all Candida species, suggesting that sexual Candida species undergo meiosis without them. Conversely, even seemingly non-mating species showed highly conserved pheromone response pathways, suggesting that pheromone signalling plays an alternative role such as regulation of biofilm formation37. These findings suggest considerable plasticity and innovation of meiotic pathways in Candida.
Moreover, we find that sexual Candida species have undergone a recent dramatic change in the pathways involved in meiotic recombination, with loss of the Dmc1-dependent pathway in the heterothallic species C. lusitaniae and C. guilliermondii (Supplementary Information, section 14c). We also found that mechanisms of chromosome pairing and crossover formation have changed recently in these two species, because they (and to a lesser extent D. hansenii) have lost several components of the synaptonemal and synapsis initiation complexes (Supplementary Table 35). They have also lost components of the major crossover-formation pathway in S. cerevisiae (MSH4, MSH5), but have retained a minor pathway (MUS81, MMS4)38,39. Overall, if Candida species undergo meiosis it is with reduced machinery, or different machinery, suggesting that unrecognized meiotic cycles may exist in many species, and that the model of meiosis developed in S. cerevisiae varies significantly, even among yeasts.
The genome sequences reported here provide a resource that will allow current knowledge of C. albicans biology, the product of decades of research, to be applied with maximum effect to the other pathogenic species in the Candida clade. They also allow many of the unusual features of C. albicans—such as cell wall gene family amplifications, and its apparent ability to undergo mating and a parasexual cycle without meiosis—to be understood in an evolutionary context that shows that the genes involved in virulence and mating have highly dynamic rates of turnover and loss.
Methods Summary
The methods for this paper are described in Supplementary Information. Here we outline the resources generated by this project.
Assemblies, gene sets, and single nucleotide polymorphisms are available in GenBank and at the Broad Institute Candida Database website (http://www.broad.mit.edu/annotation/genome/candida_group/MultiHome.html). The Broad Institute website provides search, visualization, BLAST and download of assemblies and gene sets. Gene families can be accessed by searching either for individual genes or with family identifiers (CF ). The C. parapsilosis assembly is also available at the Wellcome Trust Sanger Institute website (http://www.sanger.ac.uk/sequencing/Candida/parapsilosis/). The revised annotation of C. albicans (SC5314) is available at the Candida Genome Database (http://www.candidagenome.org).
). The C. parapsilosis assembly is also available at the Wellcome Trust Sanger Institute website (http://www.sanger.ac.uk/sequencing/Candida/parapsilosis/). The revised annotation of C. albicans (SC5314) is available at the Candida Genome Database (http://www.candidagenome.org).
Accession codes
Data deposits
Assemblies reported here have been deposited in GenBank under the following project accession numbers: AAFO00000000 (C. albicans WO-1), AAFN00000000 (C. tropicalis), AAPO00000000 (L. elongisporus), AAFM00000000 (C. guilliermondii), AAFT00000000 (C. lusitaniae), CABE01000001–CABE01000024 (C. parapsilosis).
References
Pfaller, M. A. & Diekema, D. J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20, 133–163 (2007)
Santos, M. A. & Tuite, M. F. The CUG codon is decoded in vivo as serine and not leucine in Candida albicans . Nucleic Acids Res. 23, 1481–1486 (1995)
Lockhart, S. R. et al. In Candida albicans, white-opaque switchers are homozygous for mating type. Genetics 162, 737–745 (2002)
Slutsky, B., Buffo, J. & Soll, D. R. High-frequency switching of colony morphology in Candida albicans . Science 230, 666–669 (1985)
Lockhart, S. R., Messer, S. A., Pfaller, M. A. & Diekema, D. J. Lodderomyces elongisporus masquerading as Candida parapsilosis as a cause of bloodstream infections. J. Clin. Microbiol. 46, 374–376 (2008)
Braun, B. R. et al. A human-curated annotation of the Candida albicans genome. PLoS Genet. 1, e1 (2005)
Jones, T. et al. The diploid genome sequence of Candida albicans . Proc. Natl Acad. Sci. USA 101, 7329–7334 (2004)
van het Hoog, M. et al. Assembly of the Candida albicans genome into sixteen supercontigs aligned on the eight chromosomes. Genome Biol. 8, R52 (2007)
Dujon, B. et al. Genome evolution in yeasts. Nature 430, 35–44 (2004)
Zhang, J., Hollis, R. J. & Pfaller, M. A. Variations in DNA subtype and antifungal susceptibility among clinical isolates of Candida tropicalis . Diagn. Microbiol. Infect. Dis. 27, 63–67 (1997)
Tavanti, A. et al. Population structure and properties of Candida albicans, as determined by multilocus sequence typing. J. Clin. Microbiol. 43, 5601–5613 (2005)
Chu, W. S., Magee, B. B. & Magee, P. T. Construction of an SfiI macrorestriction map of the Candida albicans genome. J. Bacteriol. 175, 6637–6651 (1993)
Forche, A., Magee, P. T., Magee, B. B. & May, G. Genome-wide single-nucleotide polymorphism map for Candida albicans . Eukaryot. Cell 3, 705–714 (2004)
Legrand, M. et al. Haplotype mapping of a diploid non-meiotic organism using existing and induced aneuploidies. PLoS Genet. 4, e1 (2008)
Massey, S. E. et al. Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res. 13, 544–557 (2003)
Bates, S. et al. Candida albicans Iff11, a secreted protein required for cell wall structure and virulence. Infect. Immun. 75, 2922–2928 (2007)
Hoyer, L. L., Green, C. B., Oh, S. H. & Zhao, X. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family–a sticky pursuit. Med. Mycol. 46, 1–15 (2008)
Yeater, K. M. et al. Temporal analysis of Candida albicans gene expression during biofilm development. Microbiology 153, 2373–2385 (2007)
Phan, Q. T. et al. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 5, e64 (2007)
Almeida, R. S. et al. The hyphal-associated adhesin and invasin Als3 of Candida albicans mediates iron acquisition from host ferritin. PLoS Pathog. 4, e1000217 (2008)
Verstrepen, K. J., Jansen, A., Lewitter, F. & Fink, G. R. Intragenic tandem repeats generate functional variability. Nature Genet. 37, 986–990 (2005)
Kaur, R., Ma, B. & Cormack, B. P. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata . Proc. Natl Acad. Sci. USA 104, 7628–7633 (2007)
Chau, A. S. et al. Inactivation of sterol Δ5,6-desaturase attenuates virulence in Candida albicans . Antimicrob. Agents Chemother. 49, 3646–3651 (2005)
Nielsen, K. & Heitman, J. Sex and virulence of human pathogenic fungi. Adv. Genet. 57, 143–173 (2007)
Noble, S. M. & Johnson, A. D. Genetics of Candida albicans, a diploid human fungal pathogen. Annu. Rev. Genet. 41, 193–211 (2007)
van der Walt, J. P. Lodderomyces, a new genus of the Saccharomycetaceae. Antonie Van Leeuwenhoek 32, 1–5 (1966)
van der Walt, J. P., Taylor, M. B. & Liebenberg, N. V. D. W. Ploidy, ascus formation and recombination in Torulaspora (Debaryomyces) hansenii . Antonie Van Leeuwenhoek 43, 205–218 (1977)
Hull, C. M., Raisner, R. M. & Johnson, A. D. Evidence for mating of the “asexual” yeast Candida albicans in a mammalian host. Science 289, 307–310 (2000)
Tsong, A. E., Miller, M. G., Raisner, R. M. & Johnson, A. D. Evolution of a combinatorial transcriptional circuit: a case study in yeasts. Cell 115, 389–399 (2003)
Tsong, A. E., Tuch, B. B., Li, H. & Johnson, A. D. Evolution of alternative transcriptional circuits with identical logic. Nature 443, 415–420 (2006)
Logue, M. E., Wong, S., Wolfe, K. H. & Butler, G. A genome sequence survey shows that the pathogenic yeast Candida parapsilosis has a defective MTLa1 allele at its mating type locus. Eukaryot. Cell 4, 1009–1017 (2005)
Fabre, E. et al. Comparative genomics in hemiascomycete yeasts: evolution of sex, silencing, and subtelomeres. Mol. Biol. Evol. 22, 856–873 (2005)
Jeffries, T. W. et al. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis . Nature Biotechnol. 25, 319–326 (2007)
Zordan, R. E., Miller, M. G., Galgoczy, D. J., Tuch, B. B. & Johnson, A. D. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans . PLoS Biol. 5, e256 (2007)
Tuch, B. B., Galgoczy, D. J., Hernday, A. D., Li, H. & Johnson, A. D. The evolution of combinatorial gene regulation in fungi. PLoS Biol. 6, e38 (2008)
Tzung, K. W. et al. Genomic evidence for a complete sexual cycle in Candida albicans . Proc. Natl Acad. Sci. USA 98, 3249–3253 (2001)
Daniels, K. J., Srikantha, T., Lockhart, S. R., Pujol, C. & Soll, D. R. Opaque cells signal white cells to form biofilms in Candida albicans . EMBO J. 25, 2240–2252 (2006)
Argueso, J. L., Wanat, J., Gemici, Z. & Alani, E. Competing crossover pathways act during meiosis in Saccharomyces cerevisiae . Genetics 168, 1805–1816 (2004)
de los Santos, T. et al. The Mus81/Mms4 endonuclease acts independently of double-Holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 164, 81–94 (2003)
Scannell, D. R., Byrne, K. P., Gordon, J. L., Wong, S. & Wolfe, K. H. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 440, 341–345 (2006)
Acknowledgements
We thank the US National Human Genome Research Institute (NHGRI) for support under the Fungal Genome Initiative at the Broad Institute. We thank C. Kurtzman for providing the sequenced strains of L. elongisporus, C. guilliermondii and C. lusitaniae, M. Koehrsen for Broad Institute website support, D. Park for informatics support and K. Wolfe and A. Regev for comments on the manuscript. We acknowledge the contributions of the Broad Institute Sequencing Platform and A. Barron, L. Clark, C. Corton, D. Ormond, D. Saunders, K. Seeger and R. Squares from the Wellcome Trust Sanger Institute for the C. parapsilosis sequencing and assembly. N.A.R.G., A.J.P.B., M.B. and co-workers were supported by the Wellcome Trust; M.G., S.S., Q.Z., C.K., B.W.B. and C.A.C. were supported by NHGRI and the National Institute of Allergy and Infectious Disease, US National Institutes of Health (NIH), Department of Health and Human Services; G.B. and co-workers were supported by Science Foundation Ireland; J.B., A.F., J.H., A.M.N. and co-workers were supported by the NIH; and M.K., M.D.R. and M.F.L. by the NIH, the US National Science Foundation and the Sloan Foundation.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Notes and Methods, Supplementary References and Supplementary Figures 1-23 with Legends (PDF 4742 kb)
Supplementary Tables
This file contains Supplementary Tables 1-35 (XLS 760 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Butler, G., Rasmussen, M., Lin, M. et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662 (2009). https://doi.org/10.1038/nature08064
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature08064
This article is cited by
-
Menthol-responsive proteome of Candida albicans
Journal of Proteins and Proteomics (2024)
-
Evolution of loss of heterozygosity patterns in hybrid genomes of Candida yeast pathogens
BMC Biology (2023)
-
Genetic Diversity of Human Fungal Pathogens
Current Clinical Microbiology Reports (2023)
-
Isolation of Clavispora lusitaniae from the Oral Cavity of Immunocompetent Young Adults from the North of Mexico
Indian Journal of Microbiology (2023)
-
Is There a Relationship Between Mating and Pathogenesis in Two Human Fungal Pathogens, Candida albicans and Candida glabrata?
Current Clinical Microbiology Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
