
Abstract
The association of genetic variation with disease and drug response, and improvements in nucleic acid technologies, have given great optimism for the impact of ‘genomic medicine’. However, the formidable size of the diploid human genome1, approximately 6 gigabases, has prevented the routine application of sequencing methods to deciphering complete individual human genomes. To realize the full potential of genomics for human health, this limitation must be overcome. Here we report the DNA sequence of a diploid genome of a single individual, James D. Watson, sequenced to 7.4-fold redundancy in two months using massively parallel sequencing in picolitre-size reaction vessels. This sequence was completed in two months at approximately one-hundredth of the cost of traditional capillary electrophoresis methods. Comparison of the sequence to the reference genome led to the identification of 3.3 million single nucleotide polymorphisms, of which 10,654 cause amino-acid substitution within the coding sequence. In addition, we accurately identified small-scale (2–40,000 base pair (bp)) insertion and deletion polymorphism as well as copy number variation resulting in the large-scale gain and loss of chromosomal segments ranging from 26,000 to 1.5 million base pairs. Overall, these results agree well with recent results of sequencing of a single individual2 by traditional methods. However, in addition to being faster and significantly less expensive, this sequencing technology avoids the arbitrary loss of genomic sequences inherent in random shotgun sequencing by bacterial cloning because it amplifies DNA in a cell-free system. As a result, we further demonstrate the acquisition of novel human sequence, including novel genes not previously identified by traditional genomic sequencing. This is the first genome sequenced by next-generation technologies. Therefore it is a pilot for the future challenges of ‘personalized genome sequencing’.
Similar content being viewed by others



Main
To catalogue the genomic diversity within a single individual, a total of 106.5 million high-quality reads were generated by 454-sequencing3, representing approximately 24.5 billion DNA bases. Reads that aligned to the genome were further filtered using stringent criteria to ensure the accuracy of mapping, resulting in 93.2 million reads aligned to reference genome sequence. The reference genome sequence was thus covered to an average depth of 7.4-fold (Fig. 1a). The alignments between the uniquely mapped reads and the reference genome were used to catalogue genetic variation in the subject’s DNA, including single nucleotide polymorphisms (SNPs), small insertions and deletions (indels), and copy number variation (CNV).

a, Distribution of sequence coverage of reference genome by 454-reads is random. Coverage, calculated in consecutive 5-kb windows, exhibited a Poisson distribution with a mean of 7.4-fold across all chromosomes except the X. Shoulder at 3.7X represents coverage of the X chromosome. b, Coverage is a key factor in detection of both alleles at heterozygous positions. For 31,709 markers heterozygous by microarray, but which exhibited only a single allele by DNA sequencing, the coverage was lower (red line, mean 5.7X) than the overall coverage for all SNPs (blue line, mean 7.8X). c, Size distribution of deletions. Deletions were readily observed in alignments of 454-reads to the reference genome. Note the peak in the size range at 300–350 bases owing to polymorphic Alu transposon insertion sites.
The 454 base-calling software provides error estimates (Q values) for each base. We developed a three-step filtering process using the patterns of error and associated Q values from the 454 base-calling software to improve the accuracy of SNP discovery. An initial 14 million variant positions were filtered to 3.32 million putative SNPs (Table 1).
Comparison of these putative SNPs in the subject’s genome with those in the dbSNP (dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/) revealed 2.72 million in common (‘known SNPs’). Approximately 99% of SNPs in dbSNP are bi-allelic. At only 10,425 positions did the subject’s variant not match the variant found in dbSNP. Although some of these could represent a third allele in the population, or an error in the dbSNP polymorphism record, we conservatively estimated the false discovery rate in the known SNPs to be approximately 0.38% based on the mismatches with dbSNP.
The remaining 0.61 million SNPs were at positions not previously identified as polymorphic in dbSNP (‘novel SNPs’). The known SNPs were divided almost equally between homozygous (50.2%) and heterozygous (49.8%) SNPs, whereas within the novel SNPs heterozygotes predominate (83.3%) compared with homozygotes (16.7%). Because most common alleles in human populations are already captured in dbSNP, novel variants are expected to be rare, and therefore much more likely to be found as heterozygotes.
We assessed the accuracy of the known SNPs derived from DNA sequencing by comparison with the experimental genotyping of the subject’s DNA using an Affymetrix 500K microarray. Compared with a haploid reference sequence, there are four possible outcomes of SNP array genotyping: homozygous for the reference allele; homozygous for the variant (non-reference) allele; heterozygous; and assay failure. Table 2 shows the results for 494,713 markers that were successfully genotyped. The subject’s DNA sequence exhibited only the reference allele at 99.4% of the markers homozygous for the reference and at 95.1% of markers homozygous for the variant. Genotyping identified 135,413 heterozygous markers of which 75.8% exhibited two alleles in the 454-reads. The lower sensitivity of detection of heterozygotes is predicted by a Poisson process of sampling DNA fragments modelled on a diploid genome (Methods). Consistent with this model, the coverage was lower at the 24.2% of heterozygous positions where DNA sequencing represented only one of the correct alleles (Fig. 1b and Supplementary Table 2). The Poisson model further shows that 13-fold average coverage would be required to detect 99% of all heterozygous SNPs (Supplementary Fig. 4).
The DNA sequencing genotypes disagree with the SNP array genotyping in 4,948 cases, or 1.0% of the time; another 3,499 markers (0.30%) had no coverage, consistent with the genome-wide redundancy of the sequence. Assuming the sensitivity and specificity of the markers on the microarray is representative of those found throughout the human genome, we estimate the total number of SNPs in the subject’s genome to be approximately 3.7 million (see ‘Sensitivity and specificity of SNP discovery’ in Supplementary Information).
We identified 222,718 indels ranging from 2 to 38,896 bp, with 113,539 in common with indels in dbSNP; 85,418 indels are found as homozygotes, and 137,300 were heterozygotes. Insertions account for 65,677 events, and deletions 157,041. A portion of the deletion size distribution from 2 to 1000 bp is shown in Fig. 1c. The distribution of deletions shows the relative enrichment for events in the range 300–350 bases in length, as expected from the known polymorphism of Alu SINE elements4,5. The size range over which insertions are detected is limited by the length of the reads; in the analysis of our 250 base-pair 454-sequencing reads, the largest observed was 208 bp.
A total of 345 indels were observed to overlap coding sequence and had the potential to alter protein function. We designed primers to amplify and validate by Sanger sequencing 111 of these events with a size range of 3–50 bases. A total of 78 indels were successfully validated of which 66 were observed to be in length multiples of 3, ranging from 3 to 33 bp, and hence not expected to cause protein translation frame shifts. Sixty-five of these indels were found as heterozygotes. Surprisingly, a 4-bp deletion in exon 11 of SGEF was found to be homozygous; however, this gene is highly conserved in vertebrate species from rhesus macaque to stickleback, and all manifest the same 4-bp deletion in their genome (Supplementary Fig. 5). Furthermore, two other independent human messenger RNAs (mRNAs) harbour the deletion as well, suggesting the subject’s allele was the wild type and that the reference harbours a rare insertion.
CNVs are local gains or losses of regions in the genome owing to duplication or deletion that can be associated with genetic disease6 and which should be detectable by variation in the average DNA sequence coverage of the region. A comparative genomic hybridization (CGH) microarray analysis of the subject’s DNA revealed 23 apparent CNV regions ranging in size from 26 kb to 1.6 Mb: 9 with DNA gains and 14 with a loss. The sequence coverage data exhibited a gain or loss congruent with the CGH result at 18 of the 23 regions (Supplementary Table 4). Regions of CNV are polymorphic in populations, segregating as alleles with varying frequency7,8. Consequently, the interpretation of a CGH microarray depends on the reference genome with which the subject is compared. This difference in reference standard is unavoidable when comparing CGH with DNA sequencing results using National Center for Biotechnology Information (NCBI) build 36 as the reference, which is not based on a single individual and for which no physical DNA sample exists. We experimentally demonstrated variation in CGH results by repeating the CGH array using a second reference genome and two different array platforms, demonstrating the effect the reference DNA has on the outcome of a CGH experiment (Supplementary Table 4).
An individual region of homozygous loss was characterized further using CGH results and DNA sequencing (Supplementary Fig. 6c). Sequence alignment of the subject’s reads spanning the breakpoint of a homozygous deletion region reveals a 2-bp addition at the breakpoint junction, suggesting non-homologous end joining6 was the mechanism involved in generating the deletion, and demonstrating the feasibility of using 454 sequence reads for identifying CNV breakpoints (Supplementary Fig. 6c). Several other CNV regions were flanked by repeats and segmental duplications, and likely occur by non-allelic homologous recombination, as was reported recently for the CNV loss at 22q13.1 (ref. 9) (see CNV 23 in Supplementary Table 4).
None of the CNV regions we defined are currently known to be involved in a recognizable phenotype; however, either trait or disease susceptibility correlations could occur in the future10. Thirty-four genes are predicted to be affected by these gains and losses, including two separate olfactory receptor groups, several genes with possible roles in cancers of the prostate, breast and colon, a gene from the HLA-D locus, and two proteins thought to be involved in RNA editing (Supplementary Table 4).
Among the 3.3 million SNPs found in the subject’s genome were 8,996 non-synonymous changes in known SNPs and 1,573 in novel SNPs. We compared the non-synonymous known SNPs with the Human Gene Mutation Database (HGMD), the largest current compendium of human disease alleles11. Thirty-two alleles exactly matched mutations reported in the HGMD whereas an additional 310 of these were in HGMD genes but were either alleles or amino-acid positions not previously characterized as disease-causing. In 12 cases the specific alterations and loci consisted of genes where homozygous recessive alleles can give rise to disease or other recognizable phenotype (Table 3); and 20 cases are reported to be associations with increased disease risk (Supplementary Table 5).
Ten of the 12 alleles in Table 3 are thought to be highly penetrant, Mendelian recessive disease-causing alleles. Seven of ten were heterozygous in the subject’s genome sequence; the other three only exhibited one allele but have an average sequence coverage less than fivefold. Because the subject does not have these three diseases, and we expect not to recover the second allele for 24% of heterozygous positions, it is likely that he is not homozygous for these disease allele positions. We note that there are not yet any systematic studies of the population frequencies of these alleles. Nevertheless, the subject is a carrier for ten highly penetrant genetic disease loci found in the HGMD data set consisting of 900 genes. It has been estimated there are fewer than ten lethal equivalents in each person12,13,14. Because we have drawn ten from an HGMD subset of the genome, we would predict the subject harbours a much greater number of deleterious Mendelian mutant alleles than is commonly estimated.
In addition, a sampling of 3,898 of the non-synonymous SNPs were tested for their possible functional impact on the protein sequence using the software Polyphen15. Polyphen classified 7.3% as ‘probably damaging’, suggesting these changes will be of functional consequence to the protein. The remainder were classified as either ‘possibly damaging’ (13%) or ‘benign’ (74%).
The genome sequence of another individual (C. Venter) was recently reported2. That study reported a 7.5-fold genome coverage using Sanger reads. The Venter genome harboured approximately 2.8 million known SNPs and about 0.74 million novel SNPs, in close agreement with the results from 454-reads of the Watson genome at a similar fold coverage. The two individuals shared 1.68 million of the SNPs, of which 5,230 were non-synonymous, accounting for 58% of the subject’s non-synonymous SNPs. Watson and Venter are each distinguished from the reference by 3,766 and 3,882 non-synonymous SNPs, respectively, and therefore are different from each other by 7,648 protein coding changes. This is the most comprehensive comparison of the non-synonymous difference between two diploid genomes yet undertaken.
The subject’s data also contained 1.5 million reads of novel sequence that did not map to build-36, corresponding to about 1.4% of the total sequence data. Approximately 65% of the unmapped sequences matched to known human repeats enriched for satellite DNA and other repeat elements characteristic of heterochromatin (Supplementary Fig. 7). The novel reads were assembled into approximately 170,000 contigs spanning 48 Mb. After removing contigs with fewer than 100 bp of contiguous unique sequence, 110,000 contigs spanning 29 Mb remained, which is close to the 25 Mb of euchromatic sequence predicted to be absent from the reference genome1. These non-repeat contigs closely match to 33 human complementary DNA (cDNA) sequences from a variety of tissues and predicted functions (Supplementary Table 6) having no known map location on the human reference genome (see Methods). To assess further the gene-coding potential of these novel DNA sequences, we compared conceptual translations of the contigs greater than 1,000 bp (1,279 in all) with the GenBank non-redundant (NR) protein database. This search yielded 60 significant, but not identical, matches to 49 different proteins in humans and other vertebrates (Supplementary Table 7). The annotations of several of these transcripts are consistent with transcription factor or signalling molecules. Therefore it is possible this diploid genome sequence will contribute important new genes to the human genome.
The sequencing method used in this study has many advantages over traditional capillary sequencing. It is inherently scalable, which means that sequencing costs in the miniature continue to decrease and throughput increases as the density of the sequencing reactions on the chip increases, and read lengths get longer3. In this study we sequenced the genome of Dr Watson for less than US$1 million, whereas the genome of Venter by Sanger sequence reportedly cost approximately US$100 million. Although not used in this study, this sequencing technology allows the production of mate-paired reads. The use of mate-pair reads will enable assessment of a wider range of indels and other structural rearrangements, and facilitate the incorporation of new sequence into the reference genome16. The principal weakness of the method is that it currently does not allow efficient detection of single-base indels in homopolymers. Future developments in chemistry and software will improve the ability to identify single-base indels.
A key aim of personal genome sequencing is to identify genome sequences that may be associated with disease, or are predictive of response to medication. The need to make genotype–phenotype correlations before having predictive value is at the heart of both the excitement and the dilemma of the new era of genomic medicine17. Thus the ability to sequence individuals readily using high-throughput, scaleable, low-cost, completely in vitro technology, as demonstrated here, is an important milestone in our ability to connect ‘personalized genomes’ to ‘personalized medicine’ and enable these critical correlations to be made.
Methods Summary
Mapping and alignment to the genome
DNA sequencing on the Genome Sequencer FLX instrument (454, Inc.) is described in detail in Methods. Reads, averaging approximately 250 bases, were mapped on to the human reference genome, NCBI build 36, by sequence alignment using Basic Local Alignment Search Tool (BLAST)-like alignment tool (BLAT). Reads were removed from subsequent analysis that failed to meet minimum criteria (see Methods). Reads that passed the alignment quality criteria were realigned to local reference genome fragments using Cross_match software; the refined alignments were parsed for sequence variation between the subject and the reference. An error model was developed to separate sequencing error from true genomic variation, and the location and type of each putative true variant was tabulated (see Methods).
Assembly of non-matching reads
One and a half million reads that failed to find a match in the reference genome sequence (‘no-hit’ reads) and another 2.2 million reads with low-quality alignments to the reference were pooled for sequence assembly. All reads were trimmed to remove the last 50 bases in which most of the sequencing error lies. Reads less than 50 bases after trimming were discarded. The assembly of the remaining 2.6 million trimmed reads followed the standard ATLAS-WGS procedure18,19.
Laboratory analysis of genomic DNA
A DNA sample from the subject was labelled and annealed to the Affymetrix 500K GeneChip array to provide independent laboratory analysis of the subject’s SNPs and conformation of the DNA sequencing coverage. To compare local fluctuation in DNA sequencing coverage with copy number variation in the subject’s genome, three additional DNA samples were labelled and mixed each in a 1:1 ratio with separately labelled control DNAs. The mixtures were annealed to each of two Agilent 244K array CGH chips and one Nimblegen HD2 chip.
Online Methods
DNA sequencing
Genomic DNA was purified from white blood cells from Dr Watson by using the Flexigene DNA kit (Qiagen). Five micrograms of DNA were sheared by nebulization and fractionated on agarose gel to isolate 450–550 base fragments. These were used to construct a single-stranded library that was used as template for single-molecule PCR on 28-μm diameter beads in emulsions3. The amplified template beads were recovered after emulsion breaking and selective enrichment. Sequencing primer was annealed to the template and the beads were incubated with Bst DNA polymerase, apyrase and single-stranded binding protein. A slurry of the template beads, enzyme beads (required for signal transduction) and packing beads (for Bst DNA polymerase retention) was loaded into the wells of a 70 mm × 75 mm picotiter plate. The picotiter plate was inserted in the flow cell and subjected to pyro-sequencing on the Genome Sequencer FLX instrument (454, Inc.).
The Genome Sequencer FLX flows 100 cycles of four solutions containing either dTTP, αSdATP, dCTP and dGTP reagents, in that order, over the cell. For each dNTP flow, a single 38-s image was captured by a CCD (charge-coupled device) camera on the sequencer. The images were processed in real time to identify template-containing wells and to compute associated signal intensities. The images were further processed for chemical and optical cross-talk, phase errors and read quality before base calling was performed for each template bead.
Mapping 454-reads to reference genome
We generated sequence with 234 runs on Genome Sequencer FLX instruments (454 Inc.), which produced over 105 million bases per run. Reads were aligned to the human reference genome, NCBI build 36 (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/bigZips/), using BLAT. All but 1.5 million reads found one or more match locations in the reference genome. The best match in the genome was used as the location for the reads with multiple matches. Poor-quality alignments were defined as those reads aligning over less than 90% of their length, or with more than four substitutions or insertions/deletions with respect to the reference, or reads that matched two locations with nearly equal match score. Ninety-three million reads comprising 7.4-fold coverage (Fig. 1a, see also Supplementary Fig. 2) passed the filtering criteria and were realigned to local genome segments (41 kb each) using Cross_match software. The limitation of a 41-kb genomic fragment placed an upper limit on the sizes of indels that could be detected.
Filtering criteria for SNPs
Mismatch base positions found among the 454-reads were scored using a scaling of the associated error probabilities (Q, see ‘Scoring system for mismatch base positions’ in Supplementary Information). The variant score, Sv, was the sum of scaled 454 Q values. Only variant positions with scores Sv ≥ 28 were considered. In addition, the ratio of variant bases to total coverage was required to be not less than 0.2.
Known SNPs were associated with homopolymer runs of more than 5 bp less than 3% of the time, whereas novel SNPs were associated 33% of the time. Therefore novel variants were removed if they were associated with a homopolymer run of more than 5 bp within 13 bases of the SNP; a novel variant was also removed if it was associated with a 5-bp homopolymer run and Sv ≤ 54 (see ‘Filtering, criteria for SNPs’ in Supplementary Information).
Genotyping with sequence data
The sensitivity of detecting heterozygous SNPs by whole-genome shotgun sequencing is limited by the depth-coverage. If the average depth-coverage is C, the coverage k for each base of the genome approximately follows the Poisson distribution21:
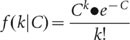
For each heterozygous SNP site of the diploid genome, covered by K reads, the number of reads i representing one of the two alleles follows the binomial distribution:
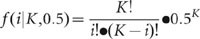
Given that a heterozygous SNP call required observation of at least two reads from both alleles at the SNP site, the sensitivity of detecting heterozygous SNPs was computed based on the two statistical distributions described above (Supplementary Fig. 4).
Filtering criteria for insertions and deletions
Indels were often associated with short tandem repeats sequences, which caused ambiguity in sequence alignments. Among separately aligned reads, indels in close proximity to one another may represent the same event. Based on our analysis of the spacing between indels, a given deletion or insertion is grouped with the previous one if: (1) the two events are the same type (insertion or deletion); (2) the ratio of the smaller to the larger is greater than or equal to 0.8; (3) the distance between the start coordinates is less than 15 bp for indels larger than 6 bp, or seven times the indel size for indels less than or equal to 6 bp. Furthermore, we required valid indels to be supported by at least two reads and have a ratio of variant to reference greater than 0.25. Errors in measurement of homopolymer length were a source of systematic error for indels one base in length, so they were ignored for this study.
Analysis of ‘no-hit’ reads
A total of 169,643 contigs were assembled with a total size of 48 Mb and an N50 size (the size at which 50% of the genome is contained within contiguous sequences of this size or greater) of 296 bp. Human repeats in the contigs were masked using RepeatMasker, and contigs containing fewer than 100 contiguous bases of unique sequence were set aside. The remaining110,353 contigs spanned 29 Mb, with an N50 size of 267 bp; 1,294 contigs were longer than 1,000 bp, and the longest was 10,724 bp.
We used Mega BLAST (http://www.ncbi.nlm.nih.gov/blast/megablast.shtml) with the ‘expect’ parameter set to 10-30 to compare these contigs to a human mRNA sequence database of over 40,000 sequences (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/bigZips/mrna.fa.gz). Significant matches were found to 417 mRNA sequences, which had no map coordinates on build 36. We selected from this set 104 mRNA that matched 886 contigs with greater than 96% identity, and which were covered across more than 40% of the mRNA length. The contigs matching these mRNA sequences ranged in size from 296 to 5121 bp. The 104 mRNA sequences were compared with the reference genome using BLAT to confirm they did not have a matching gene on the reference genome. Forty-four were eliminated by this test; 27 had a partial hit, but the genomic match and the contig match did not overlap on the mRNA sequence; 33 of the mRNA sequences had no hit.
Comparative genome hybridization
For the Agilent 244K array, the Human Genome CGH 244K Array (Agilent Technologies, Inc.) contains 238,459 formatted 60-base oligonucleotides, representing a compiled view of the human genome at an average resolution of 9 kb. DNA digestion, labelling and hybridization were performed according to the manufacturer’s instructions, with minor modifications. Two separate experiments differed in the reference DNA used for comparison: ‘Agilent_1’ used a standard reference caucasian male (Kleberg Cytogenetics Laboratory, Baylor College of Medicine); ‘Agilent_2’ used a caucasian male, NA10851, Coriell Institute for Medical Research. For the Nimblegen HD2 array, we tested a second sample of experimental DNA co-annealed with reference DNA, Coriell Institute for Medical Research number NA10851, to a NimbleChip HD2 Array (NimbleGen Systems, Inc.) in collaboration with NimbleGen Systems. The HD2 array has 2.1 million oligonucleotides, each between 50 and 75 bases, with a reported resolution of about 5 kb. Log2 ratios were analysed for variation in copy number using NimbleGen SignalMap software.
Affymetrix Gene Chip 500
Duplicate genomic DNA samples from lymphocytes, 250 ng each, were annealed to the Affymetrix 250K NspI and 250K StyI arrays according to the manufacturer’s protocol (see ‘Affymetrix Gene Chip 500’ in Supplementary Information for further details).
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The CEL files for the Affymetrix 500K Genome array and GPR files for the Agilent_1 and Agilent_2 CGH arrays are deposited in Gene Expression Omnibus under series accession number GSE10668. All SNPs and insertion/deletions from 454-reads are deposited in dbSNP under handle bcmhgsc_jdw; contigs from the assembly of 454-reads not matching the reference genome are deposited in Genbank under accession number ABKV01000000. All 454-reads are deposited in the Trace Archive of the National Center for Biotechnology Information under Center_name = ‘CSHL’ and Center_project = ‘Project JIM’.
References
The International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004)
Levy, S. et al. The diploid genome sequence of a single individual. PLoS Biol. 5, e254–e286 (2007)
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005)
Bennett, E. A., Coleman, L. E., Tsui, C., Pittard, W. S. & Devine, S. E. Natural genetic variation caused by transposable elements in humans. Genetics 168, 933–951 (2004)
The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005)
Lupski, J. R. & Stankiewicz, P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genetics 1, 627–633 (2005)
Weber, J. L. et al. Human diallelic insertion/deletion polymorphisms. Am. J. Hum. Genet. 71, 854–862 (2002)
Redon, R. et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006)
Kidd, J. M., Newman, T. L., Tuzun, E., Kaul, R. & Eichler, E. E. Population stratification of a common APOBEC gene deletion polymorphism. PLOS Genetics 3, 584–592 (2007)
Lupski, J. R. Structural variation in the human genome. N. Engl. J. Med. 356, 1169–1171 (2007)
Stenson, P. D. et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581 (2003)
Halligan, D. L. & Keightley, P. D. How many lethal alleles? Trends Genet. 19, 57–59 (2003)
Bittles, A. H. & Neel, J. V. The costs of human inbreeding and their implications for variations at the DNA level. Nature Genet. 8, 117–121 (1994)
Vogel, F. & Motulsky, A. G. Human Genetics: Problems and Approaches 2nd edn 487–502 (Springer-Verlag, New York, 1986)
Ramensky, V., Bork, P. & Sunyaev, S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900 (2002)
Korbel, J. O. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426 (2007)
McGuire, A. L., Caulfield, T. & Cho, M. K. Research ethics and the challenge of whole genome sequencing. Nature Rev. Genet. 9, 152–156 (2008)
Havlak, P. et al. The Atlas genome assembly system. Genome Res. 14, 721–732 (2004)
Richards, S. et al. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 15, 1–18 (2005)
Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 (1998)
Lander, E. S. & Waterman, M. S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2, 231–239 (1988)
Acknowledgements
We thank J. D. Watson for his participation, encouragement and engagement. We thank J. Belmont for discussion of HGMD data. We also thank the following individuals for their technical support: S. Attiya, M. Braverman, J. Brunelle, C. Celone, Z. Chen, A. Sancher, W. Song, and personnel from 454 Sequencing Center and R&D.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
The file contains extensive Supplementary Information with Supplementary Figures 1-15. (PDF 3488 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Wheeler, D., Srinivasan, M., Egholm, M. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–876 (2008). https://doi.org/10.1038/nature06884
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature06884
This article is cited by
-
The reckoning of chromosomal instability: past, present, future
Chromosome Research (2024)
-
30 years of progress from positional cloning to precision genome editing
Nature Genetics (2022)
-
Temporal dynamics of teleost populations during the Pleistocene: a report from publicly available genome data
BMC Genomics (2021)
-
Unexpected organellar locations of ESCRT machinery in Giardia intestinalis and complex evolutionary dynamics spanning the transition to parasitism in the lineage Fornicata
BMC Biology (2021)
-
Automated Flow Synthesis of Tumor Neoantigen Peptides for Personalized Immunotherapy
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
