
Abstract
Bacillus cereus is an opportunistic pathogen causing food poisoning manifested by diarrhoeal or emetic syndromes1. It is closely related to the animal and human pathogen Bacillus anthracis and the insect pathogen Bacillus thuringiensis, the former being used as a biological weapon and the latter as a pesticide. B. anthracis and B. thuringiensis are readily distinguished from B. cereus by the presence of plasmid-borne specific toxins (B. anthracis and B. thuringiensis) and capsule (B. anthracis). But phylogenetic studies based on the analysis of chromosomal genes bring controversial results, and it is unclear whether B. cereus, B. anthracis and B. thuringiensis are varieties of the same species2 or different species3,4. Here we report the sequencing and analysis of the type strain B. cereus ATCC 14579. The complete genome sequence of B. cereus ATCC 14579 together with the gapped genome of B. anthracis A20125 enables us to perform comparative analysis, and hence to identify the genes that are conserved between B. cereus and B. anthracis, and the genes that are unique for each species. We use the former to clarify the phylogeny of the cereus group, and the latter to determine plasmid-independent species-specific markers.
Similar content being viewed by others

Main
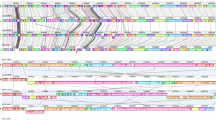
The general features of the B. cereus ATCC 14579 genome are listed in Table 1. The region between 0.8 and 1.8 megabases (Mb) has G + C content close to the average for the chromosome (35.3%); in the region between 3.7 and 0.8 Mb the G + C content is higher than the average value, and in the region between 1.8 and 3.7 Mb it is lower than the average value (Fig. 1, circle 3). These regions are bordered with putative prophages (Fig. 1, circles 6 and 7), which could be indicative of the origin of the B. cereus ATCC 14579 chromosome as a result of phage-mediated recombination between the chromosomes of closely related bacteria.

From the inside: green and blue bars show positions of two insertion sequence (IS) elements, red-to-black bars show positions of rrn operons. Circle 1, G + C content over 200-kb window with 5-kb step; red and blue denote respectively G + C content higher and lower than average. Circles 2 and 3, G + C and (C - G)/(C + G) content over 20-kb window with 5-kb step. Circles 4 and 5, CDSs on the - and + strands, colour reflects functional category (see key). Circle 6, homology to B. subtilis CDSs using FASTA. Green, 0–30% identity; blue, 30–45%; red, 45–65%; pink, 65–100%.
A 15.1-kilobase (kb) contig with G + C content of 38%, which could not be joined to any other by the multiplex long accurate (MLA) polymerase chain reaction (PCR) procedure or assembled into a separate circular structure by long-range (LR) PCR, corresponds to the linear plasmid, detected earlier in total DNA preparations from this bacterium6. One of the coding sequences (CDSs) in this contig is homologous to a type B DNA polymerase typically found in Bacillus subtilis phage Φ29 and several linear mitochondrial plasmids. The contig also contains a CDS with similarity to endolysin. We therefore suggest that it corresponds to a linear plasmid or prophage, which we designated pBClin15. The termini of the plasmid could be protected by covalently closed hairpin telomeres or by a covalently bound protein, as in the phage Φ29. However, pBClin15 carries no CDS with homology to the phage terminal protein. The polymerase gene of pBClin15 is more closely related to those of mitochondrial linear plasmids than to polymerases of B. subtilis phages.
Phylogenetic analysis of the cereus group2,3,4 shows that B. cereus ATCC 14579 and B. anthracis strains are not particularly close, and the ecological niches occupied by these bacteria are also very different7. Nevertheless 4,505 CDSs in B. cereus ATCC 14579 have 80–100% identity to their homologues in B. anthracis A2012. The average identity for this group of genes is 92.1%. Some general statistics of the genome comparison are also provided in Table 1 (rows 10–14). Potential orthologues identified as bidirectional best hits represent approximately 75% of the CDSs for each of the genomes. 85% of these potential orthologues have conserved neighbourhood, suggesting extensive short-range synteny (Table 1).
The large core set of genes (75–80%) conserved between B. cereus ATCC 14579 and B. anthracis A2012 could have been inherited from a common ancestor. Analysis of the metabolic potential encoded by the core set contradicts the hypothesis of the cereus group common ancestor being a soil bacterium. The characteristic feature of soil bacteria, such as Streptomyces spp. or B. subtilis, is the multiplicity of carbohydrate catabolic pathways reflecting the variety of carbohydrates in the soil, where plant-derived material is the main source of nutrients. Whereas a total of 41 genes for degradation of carbohydrate polymers were identified in the B. subtilis genome, only 14 and 15 CDSs coding for polysaccharide degradation enzymes are present in B. cereus and B. anthracis, respectively, and the spectrum of polysaccharides that can be degraded by these bacteria is limited to glycogen and starch, chitin and chitosan (Supplementary Fig. 1). In contrast, the abundance of proteolytic enzymes, the multiplicity of peptide and amino-acid transporters and the variety of amino-acid degradation pathways (Supplementary Table 1) indicates that proteins, peptides and amino acids may be a preferred nutrient source for B. cereus and B. anthracis. A total of 51 and 48 protease-encoding CDSs were identified in B. cereus and B. anthracis, respectively, compared to only 30 in B. subtilis. While three of the B. subtilis extracellular proteases are absent from B. cereus (Epr, Bpr and AprX), other proteases, which are found in one or two copies in B. subtilis, are represented as large families in B. cereus and B. anthracis (Supplementary Table 1). Several observations suggest that the insect intestine could have been the natural habitat for the common ancestor of the cereus group8. The peritrophic membrane of insect guts consists of chitin and protein components. While the chitinolytic enzymes enable B. cereus and B. anthracis to degrade the chitin component, homologues of the zinc metalloprotease enhancin9 of entomopathogenic viruses (BC3384 and BA_3939) could provide the ability to cleave the invertebrate intestinal mucin, which is the major protein component of the insect peritrophic membrane. A potential PlcR-binding site was found upstream of the enhancin homologue in B. cereus.
The core set of genes conserved between B. cereus and B. anthracis includes numerous factors for invasion, establishment and propagation of bacteria within the host. While the presence of such genes in B. anthracis is not surprising, finding of pathogenicity-related genes in B. cereus ATCC 14579 was somewhat unexpected. Genes encoding all but two toxins ever identified in B. cereus clinical isolates were found in the reputedly non-pathogenic B. cereus ATCC 14579 (Supplementary Table 2). Both B. anthracis and B. cereus implement several mechanisms of protection against the host defence system. Three homologues of the immune inhibitor A protein (InhA), which selectively cleaves insect antibacterial peptides10, were found in B. cereus ATCC 14579, and two homologues of InhA are present in B. anthracis A2012. A homologue of staphylococcal MprF protein11 found in B. cereus (BC1465) and B. anthracis (BA_2009) could further enhance resistance to antibacterial peptides by decreasing the negative charge of the bacterial surface via aminoacylation of anionic phospholipids with lysine. The counterparts of the S-layer proteins from B. anthracis and B. thuringiensis were not found in the B. cereus ATCC 14579 genome, although several CDSs with SLH domains were identified. This observation is in agreement with the results of Kotiranta et al.12, who demonstrated that, unlike many clinical isolates, the strain ATCC 14579 is devoid of an S-layer. The presence of CDSs encoding potential pathogenicity factors in B. cereus, B. anthracis and B. thuringiensis13,14 is consistent with the cereus group ancestor being an opportunistic insect pathogen rather than a benign soil bacterium.
The repair of ultraviolet (UV)-induced DNA damage could have been of critical importance to the cereus group ancestor, as there are numerous mechanisms to repair UV-induced lesions. Direct reversal of UV-damage is mediated by two photolyases: one is spore-specific (splB-type) and the other is of the phrB-type. B. cereus utilizes a bacterial uvrABC repair system for UV-dimer excision, and is also capable of transcription-coupled repair (BC0058). In addition, B. cereus has two CDSs (BC0260, BC5347) homologous to the C-terminal endonuclease domain of the UvdE UV-repair endonuclease from Neurospora crassa and Uve1 from Schizosaccharomyces pombe.
The pleiotropic regulator PlcR was previously identified as one of the principal regulators of B. cereus virulence genes13,14,15. When no mismatches were allowed, a total of 55 possible PlcR-binding sites were found, of which 26 coincide with the promoter regions of genes and 24 were found in the upstream regions of potential operons, bringing the number of genes that could be controlled directly by PlcR to more than 100 (Supplementary Table 3). In addition to PlcR (BC5350), at least four transcriptional regulators (BC1715, BC2410, BC2770 and BC3194) belong to the potential PlcR regulon (Fig. 2), suggesting that other CDSs could be regulated by PlcR indirectly, which is in accord with recent proteomics data16. The presence of three PlcR paralogues (BC0988, BC1158 and BC2443) in the genome of B. cereus ATCC 14579 could make the PlcR regulatory network even more complex. Though none of the PlcR paralogues appears to be controlled directly by PlcR (Supplementary Table 3), it is possible that they are activated by the PapR peptide, as was demonstrated for PlcR17.

Genes with putative PlcR-binding sites in the upstream regions were classified into several categories, including toxins, proteases, surface proteins, motility and chemotaxis genes, antibiotic efflux proteins, and so on. Activation of the PlcR regulon results in secretion of numerous proteins with cytotoxic activities towards mammalian and insect cells. Expression of the plcR gene is activated by an unknown mechanism, which includes oligopeptide permease-dependent uptake of a PapR peptide encoded by CDS2 located downstream from the PlcR gene.
The histidine protein kinase (BC3528) homologous to the B. subtilis sporulation kinase KinB appears to be an important member of the PlcR regulatory network. It is known that the phosphorylated form of the transition state regulator Spo0A acts as a repressor of PlcR18, so upregulation of BC3528 by PlcR would provide a feedback mechanism that controls PlcR expression. An orthologue of protein kinase BC3528 is absent from B. anthracis A2012, which could contribute to the incompatibility of PlcR and AtxA regulons in B. anthracis19. Another potential member of the PlcR regulon, the mutator DNA polymerase IV (BC4142), could provide irreversible pathogenic adaptation of B. cereus and B. anthracis. DNA polymerase IV belongs to the DinB/UmuCD/Rad30/Rev1 superfamily of error-prone DNA polymerases that have been shown to induce adaptive mutability in bacteria under stressful conditions20. We suggest that during host colonization, polymerase IV-dependent mutagenesis could result in adaptive point mutations that enhance survival of B. cereus and related bacteria, and potentially affect their pathogenicity.
Approximately 15% of the CDSs in either genome show no similarity with the other genome (Table 1). Prophage proteins and transposases account for approximately 140 unique CDSs in each organism. We detected six prophages in the genome of ATCC 14579 that were not previously characterized in the B. cereus group. The prophages are not inserted within the transfer RNA operons, and they do not carry any of the known pathogenicity factors of B. cereus.
Some of the unique CDSs found in B. cereus ATCC 14579 and B. anthracis A2012 appear to be specific for a certain group of strains rather than species-specific. In B. cereus ATCC 14579, a chromosomal cluster that could code for capsular polysaccharide biosynthesis was found. It covers more than 20 kb (BC5279–BC5263), and contains genes for glycosyltransferases and flippase-type translocase, as well as polysaccharide polymerization machinery including chain length regulator and its regulatory protein-tyrosine kinase and phosphatase. A cluster specific for B. anthracis A2012 (BA_0356 to BA_0371), which contains several genes for glycosyltransferases and an ABC transporter similar to the teichoic acid exporter tagGH of B. subtilis, could code for biosynthesis of a teichuronic acid or a secondary cell wall polymer that serves as an anchor for S-layer proteins.
B. cereus ATCC 14579 has an extensive repertoire of restriction-modification systems, which includes type I, II and 5-methylcytosine-specific systems. The B. cereus type I system comprises restriction, methylation and specificity subunits. The type II system has restriction and methylation domains; the type II R domains are weakly similar to McrB-type and Lactococcus lactis LlaI restriction systems. B. anthracis lacks the type I and II restriction-modification systems, but, unlike B. cereus, it has a CDS weakly similar to the 5-methylcytosine-specific Mrr endonuclease.
A chromosomal cluster (BC5090–BC5083) potentially coding for biosynthesis of a novel peptide antibiotic was found in B. cereus ATCC 14579, but not in B. anthracis. The cluster includes the precursor peptide and putative modification proteins, including a homologue of the subtilin biosynthesis protein SpaB (BC5084) that catalyses dehydration of serine and threonine. However, no homologue of the thioether-forming SpaC protein was found, suggesting an unusual structure for this peptide antibiotic.
Other CDSs identified as unique for B. cereus ATCC 14579 and B. anthracis A2012 could be used as plasmid-independent species-specific markers. A seven-gene chromosomal cluster for inositol degradation is present in B. anthracis, but not in B. cereus. Compared to the B. subtilis iol operon, two genes, iolH and iolE, are missing from the B. anthracis cluster, which probably makes the B. anthracis operon non-functional, as all iol genes except iolS are essential for inositol utilization in B. subtilis21. In B. cereus, a chromosomal cluster encoding the enzymes from the arginine deiminase pathway (BC0406–BC0409) was identified. This pathway enables Streptococcus pyogenes to survive acidic conditions in the presence of arginine owing to release of ammonium22, and it may play a similar role in B. cereus. In B. anthracis, the entire arginine deiminase cluster appears to be deleted (Fig. 3), though the neighbouring CDSs are conserved. Ammonium inhibits receptor-mediated internalization of the lethal toxin23, therefore ammonium production by arginine deiminase could be disadvantageous for B. anthracis. Deletion of the arginine deiminase operon in B. anthracis could be a case of disposal of genes detrimental for the pathogenic lifestyle, similar to that of Escherichia coli lysine decarboxylase24.

The top portion of the figure shows CDSs within the equivalent genomic regions from B. anthracis and B. cereus. The orthologues of CDSs (shown as arrows of the same colour) flanking the arginine deiminase operon in B. cereus are adjacent to each other in the B. anthracis genome. Below are the DNA sequences of B. anthracis and B. cereus flanking the deletion site. Conserved nucleotides are shown red.
The availability of both complete and gapped genome sequence data from bacteria belonging to the cereus group provides a basis for whole-genome-based phylogenetic analysis, with a view to exploring the genetic diversity within the cereus group, and inferring the nature and origin of the species. These sequence data should also facilitate the identification of genes that are crucial for host colonization and bacterial propagation during B. cereus infection; such findings would have implications for understanding the biology of B. anthracis.
Methods
B. cereus strain ATCC 14579 was obtained from ATCC, and used to construct libraries in plasmids (2–3-kb inserts in pGEM3) and cosmids (30–35-kb inserts using Lorist 6). The strain 6A5, which is considered to be the same as ATCC 14579, was obtained from BGSC, and used in Génétique Microbienne, INRA, to perform LR PCR for the final stages of the project. High-molecular-mass genomic DNA was isolated from B. cereus following standard protocols. The DNA was either used for LR PCR, or sheared, size-fractionated, and used to construct libraries. Whole-genome shotgun sequencing was performed on about 30,000 plasmids and 3,000 cosmids using Applied Biosystems 3700 DNA sequencers (Perkin-Elmer). In the first phase of sequencing, the genome was assembled using Phred-Phrap-Consed into 547 contigs longer than 2,000 base pairs (bp), the longest being 50,451 bp. Gaps were closed by primer walking over cosmid clone inserts (3,000 reactions, resulting in 128 contigs longer than 2,000 bp, the longest being 273,559 bp) and by sequencing of LR PCR products using the finishing strategy based on MLA PCR25 (2,320 reactions). Finally, the genome was assembled into two contigs representing the circular chromosome and the linear plasmid with an average of 6 × coverage. The presence of a linear plasmid was confirmed by Southern hybridization. The statistical distribution of random readings between plasmid and chromosomal DNA indicates that the plasmid is present in one copy per chromosome (130 reads for the 15-kb plasmid compared to 47,315 reads for the 5,412-kb chromosome in one of the assembly versions).
The number of rrn operons was determined by LR PCR and Southern hybridization (not shown), and each operon was sequenced by primer walking. Thirteen rrn operons were found (Fig. 1), this number being higher than 5 to 12 operons reported earlier for strains of B. cereus or Bacillus weihenstephanensis26,27. The sequences of 16S, 23S and 5S ribosomal RNAs are very similar in all operons, the number of differences being up to 2, 7 and 4 bases, respectively. No 16S rRNA gene containing the psychrotolerance (ability to grow at low temperature) signature was detected. The corresponding rrn operons of B. anthracis A2012 were omitted from the deposited sequence, so no comparison could be made at this point.
Genome sequence data for B. anthracis A2012 (NC_003995) was obtained from NCBI. Genes were identified by combination of Critica and a CDS-calling program developed at Integrated Genomics (IG). Nine additional CDSs on the chromosome of B. anthracis were identified, compared to the annotation provided by NCBI, by searching intergenic DNA sequences against the IG genome database. The genomes of B. cereus and B. anthracis were subjected to a round of automatic annotation followed by extensive manual curation within the ERGO bioinformatics suite28. Comparative analysis of B. cereus and B. anthracis genomes was carried out with the WorkBench algorithm (Integrated Genomics), as described for three strains of Xylella fastidiosa29. As the sequence of B. anthracis chromosome is incomplete, the CDSs identified as unique for B. cereus were subjected to further analysis, including analysis of the chromosomal context and search for homologues in the unfinished B. anthracis and B. cereus genomes in the TIGR database (http://www.tigr.org) and in the gapped genome of B. thuringiensis israelensis30. ERGO tools for pattern matching were used to identify the genes potentially controlled by the virulence regulator PlcR13,14,15 based on the finding of the consensus sequence TATGnAnnnnTnCATA15 in the upstream region of the genes. The results of analysis, including annotations and partial metabolic reconstructions of B. anthracis A2012 and B. cereus ATCC 14579 can be found in the limited version of ERGO at http://www.ergo-light.com.
References
Kotiranta, A., Lounatmaa, K. & Haapsalo, M. Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2, 189–198 (2000)
Helgason, E. et al. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis—one species on the basis of genetic evidence. Appl. Environ. Microbiol. 66, 2627–2630 (2000)
Ticknor, L. O. et al. Fluorescent amplified fragment length polymorphism analysis of norwegian Bacillus cereus and Bacillus thuringiensis soil isolates. Appl. Environ. Microbiol. 67, 4863–4973 (2001)
Vilas-Boas, G., Sanchis, V., Lereclus, D., Lemos, M. V. F. & Bourguet, D. Genetic differentiation between sympatric populations of Bacillus cereus and Bacillus thuringiensis. Appl. Environ. Microbiol. 68, 1414–1424 (2002)
Read, T. D. et al. Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 296, 2028–2033 (2002)
Carlson, C. R., Grønstad, A. & Kolstø, A. B. Physical map of the genomes of three Bacillus cereus strains. J. Bacteriol. 174, 3750–3756 (1992)
Turnbull, P. C. B. Definitive identification of Bacillus anthracis—a review. J. Appl. Microbiol. 87, 237–240 (1999)
Margulis, L. et al. The Arthromitus stage of Bacillus cereus: intestinal symbionts of animals. Proc. Natl Acad. Sci. USA 95, 1236–1241 (1998)
Wang, P. & Granados, R. R. An intestinal mucin is the target substrate for a baculovirus enhancin. Proc. Natl Acad. Sci. USA 94, 6977–6982 (1997)
Fedhila, S., Nel, P. & Lereclus, D. The InhA2 metalloprotease of Bacillus thuringiensis strain 407 is required for pathogenicity in insects infected via the oral route. J. Bacteriol. 184, 3296–3304 (2002)
Peschel, A. et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L-lysine. J. Exp. Med. 193, 1067–1076 (2001)
Kotiranta, A. et al. Surface structure, hydrophobicity, phagocytosis, and adherence to matrix proteins of Bacillus cereus cells with and without the crystalline surface protein layer. Infect. Immun. 66, 4895–4902 (1998)
Agaisse, H., Gominet, M., Økstad, O. A., Kolstø, A. B. & Lereclus, D. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol. Microbiol. 32, 1043–1053 (1999)
Salamitou, S. et al. The plcR regulon is involved in the opportunistic properties of Bacillus thuringiensis and Bacillus cereus in mice and insects. Microbiology 146, 2825–2832 (2000)
Økstad, O. A. et al. Sequence analysis of three Bacillus cereus loci carrying PlcR-regulated genes encoding degradative enzymes and enterotoxin. Microbiology 145, 3129–3138 (1999)
Gohar, M. et al. Two-dimensional electrophoresis analysis of the extracellular proteome of Bacillus cereus reveals the importance of the PlcR regulon. Proteomics 2, 784–791 (2002)
Slamti, L. & Lereclus, D. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J. 21, 4550–4559 (2002)
Lereclus, D., Agaisse, H., Grandvalet, C., Salamitou, S. & Gominet, M. Regulation of toxin and virulence gene transcription in Bacillus thuringiensis. Int. J. Med. Microbiol. 290, 295–299 (2000)
Mignot, T. et al. The incompatibility between the PlcR- and AtxA-controlled regulons may have selected a nonsense mutation in Bacillus anthracis. Mol. Microbiol. 42, 1189–1198 (2001)
McKenzie, G. J., Lee, P. L., Lombardo, M.-J., Hastings, P. J. & Rosenberg, S. M. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol. Cell 7, 571–579 (2001)
Yoshida, K.-I., Aoyama, D., Ishio, I., Shibayama, T. & Fujita, Y. Organization and transcription of the myo-inositol operon, iol, of Bacillus subtilis. J. Bacteriol. 179, 4591–4598 (1997)
Degnan, B. A. et al. Characterization of an isogenic mutant of Streptococcus pyogenes Manfredo lacking the ability to make streptococcal acid glycoprotein. Infect. Immun. 68, 2441–2448 (2000)
Gordon, V. M., Leppla, S. H. & Hewlett, E. L. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 56, 1066–1069 (1988)
Maurelli, A. T., Fernandez, R. E., Bloch, C. A., Rode, C. K. & Fasano, A. “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl Acad. Sci. USA 95, 3943–3948 (1998)
Sorokin, A. et al. A new approach using multiplex long accurate PCR and yeast artificial chromosomes for bacterial chromosome mapping and sequencing. Genome Res. 6, 448–453 (1996)
Johansen, T., Carlson, C. R. & Kolstø, A. B. Variable number of rRNA operons in Bacillus cereus strains. FEMS Microbiol. Lett. 136, 325–328 (1996)
Lechner, S. et al. Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int. J. Syst. Bacteriol. 48, 1373–1382 (1998)
Kapatral, V. et al. Genome sequence and analysis of the oral bacterium Fusobacterium nucleatum strain ATCC 25586. J. Bacteriol. 184, 2005–2018 (2002)
Bhattacharyya, A. et al. Whole-genome comparative analysis of three phytopathogenic Xylella fastidiosa strains. Proc. Natl Acad. Sci. USA 99, 12403–12408 (2002)
Anderson, I. et al. Genome of Bacillus thuringiensis subsp. israelensis and comparative genomics of the Bacillus cereus group. J. Bacteriol (submitted)
Acknowledgements
This Letter is dedicated to the memory of our dear colleagues M. Mazur and C. Anagnostopoulos. This work was supported by a DARPA STTR grant to Integrated Genomics Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Ivanova, N., Sorokin, A., Anderson, I. et al. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 423, 87–91 (2003). https://doi.org/10.1038/nature01582
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature01582
This article is cited by
-
Impacts of UV radiation on Bacillus biocontrol agents and their resistance mechanisms
World Journal of Microbiology and Biotechnology (2024)
-
De novo assembly and comparative genome analysis for polyhydroxyalkanoates-producing Bacillus sp. BNPI-92 strain
Journal of Genetic Engineering and Biotechnology (2023)
-
Genomic analysis and assessment of pathogenic (toxicogenic) potential of Staphylococcus haemolyticus and Bacillus paranthracis consortia isolated from bovine mastitis in Russia
Scientific Reports (2023)
-
Identification of a novel cyclomaltodextrinase annotated as a neopullulanase in the genome of Bacillus cereus
Archives of Microbiology (2023)
-
Characterization of bacterial communities responsible for bioplastics degradation during the thermophilic and the maturation phases of composting
Journal of Material Cycles and Waste Management (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
