
Abstract
The epigenome is associated with biological factors, such as disease status, and environmental factors, such as smoking, alcohol consumption and body mass index. Although there is a widespread perception that environmental influences on the epigenome are pervasive and profound, there has been little evidence to date in humans with respect to environmental factors that are biologically distal. Here we provide evidence on the associations between epigenetic modifications—in our case, CpG methylation—and educational attainment (EA), a biologically distal environmental factor that is arguably among the most important life-shaping experiences for individuals. Specifically, we report the results of an epigenome-wide association study meta-analysis of EA based on data from 27 cohort studies with a total of 10 767 individuals. We find nine CpG probes significantly associated with EA. However, robustness analyses show that all nine probes have previously been found to be associated with smoking. Only two associations remain when we perform a sensitivity analysis in the subset of never-smokers, and these two probes are known to be strongly associated with maternal smoking during pregnancy, and thus their association with EA could be due to correlation between EA and maternal smoking. Moreover, the effect sizes of the associations with EA are far smaller than the known associations with the biologically proximal environmental factors alcohol consumption, body mass index, smoking and maternal smoking during pregnancy. Follow-up analyses that combine the effects of many probes also point to small methylation associations with EA that are highly correlated with the combined effects of smoking. If our findings regarding EA can be generalized to other biologically distal environmental factors, then they cast doubt on the hypothesis that such factors have large effects on the epigenome.
Similar content being viewed by others
Introduction
The epigenome has been shown to be associated with biological factors such as disease status.1, 2 Although there is a widespread perception in the social sciences that a variety of social environmental factors have an effect on the epigenome,3, 4, 5, 6, 7, 8, 9, 10 virtually all of the replicated evidence to date in humans relates to environmental factors that have a fairly direct biological impact, such as smoking,11, 12, 13 alcohol consumption14, 15 and excess energy intake resulting in increased body mass index (BMI).16, 17 Here we study the associations between epigenetic modifications—specifically, the methylation of cytosine–guanine pairs connected by a phosphate link (CpG methylation)—and educational attainment (EA). EA is biologically distal, and yet it is arguably among the most important life-shaping experiences for individuals in modern societies. EA therefore provides a useful test case for whether and to what extent biologically distal environmental factors may affect the epigenome.
In this paper, we report the results of a large-scale epigenome-wide association study (EWAS) meta-analysis of EA. By meta-analyzing harmonized EWAS results across 27 cohort studies, we were able to attain an overall sample size of 10 767 individuals of recent European ancestry, making this study one of the largest EWAS to date.13, 15, 18 A large sample size is important because little is known about plausible EWAS effect sizes for complex phenotypes such as EA, and an underpowered analysis would run a high risk of both false negatives and false positives.19, 20
As is standard in EWAS, we used data on CpG DNA methylation. This is the most widely studied epigenetic mark in large cohort studies.1 Methylation level was measured by the beta value, which is the proportion of methylated molecules at each CpG locus, a continuous variable ranging between 0 and 1.21 The Illumina 450k Bead Chip measures methylation levels at >480 000 loci in human DNA and has been used in many cohort studies.1
We report results from two common methods for the analysis of such methylation data sets. The first main analysis is an EWAS, which considers regression models for each CpG locus with EA. Using the EWAS results, we then performed a series of follow-up analyses: enrichment analyses, prediction analyses, correlation with tissue-specific methylation, and gene-expression analysis (Supplementary Note). The second main analysis uses the ‘epigenetic clock’22, 23 method, which employs a weighted linear combination of a subset of probes (that is, measured CpG methylation loci) to predict an individual’s so-called ‘biological age’. The resulting variable can then be linked to phenotypes and health outcomes.
EWAS studies to date have found associations between DNA methylation and, for example, smoking,11, 12 BMI,16, 24 traumatic stress,25 alcohol consumption14, 26 and cancer.2, 27 In prior work, an age-accelerated epigenetic clock (that is, an older biological than chronological age) has been linked to increased mortality risk,28 poorer cognitive and physical health,29 greater Alzheimer’s disease pathology,30 Down’s syndrome,31 high lifetime stress32 and lower income.33
Methods
Participating cohorts
We obtained summary-level association statistics from 27 independent cohort studies across 15 cohorts located in Europe, the United States and Australia (Supplementary Table S1.1). The total sample size comprised 10 767 individuals of recent European ancestry. All participants provided written informed consent, and all contributing cohorts confirmed compliance with their Local Research Ethics Committees or Institutional Review Boards.
EA measures
Following earlier work of the Social Science Genetic Association Consortium,34, 35 EA was harmonized across cohorts. The EA variable is defined in accordance with the ISCED 1997 classification (UNESCO), leading to seven categories of EA that are internationally comparable. The categories are translated into US years-of-schooling equivalents, which have a quantitative interpretation (Supplementary Tables S1.2 and S1.3).
Participant inclusion criteria
To be included in the current analysis, participants had to satisfy six criteria: (1) participants were assessed for EA at or after 25 years of age; (2) participants were of European ancestry; (3) all relevant covariate data were available for each participant; (4) participants passed the cohort-level methylation quality control (QC); (5) participants passed cohort-specific standard QCs (for example, genetic outliers were excluded); and (6) participants were not disease cases from a case/control study.
DNA methylation measurement and cohort-level QC
Whole-blood DNA CpG methylation was measured genome-wide in all cohorts using the Illumina 450k Human Methylation chip (San Diego, CA, USA). We standardized the cohort-level QC and preprocessing of the methylation data as much as possible, while ensuring some degree of flexibility to keep the implementation feasible for all cohorts (leading to slight variation in preprocessing across cohorts, as is common13, 15, 17). Cohort-specific information regarding technical treatment of the data, such as background-correction,36 normalization37 and QC, is reported in Supplementary Table S1.4.
Statistical analysis
Our analyses were performed in accordance with a preregistered analysis plan archived at Open Science Framework (OSF) in September 2015 (available at: https://osf.io/9v3nk/).
Epigenome-wide association study
To investigate associations with individual methylation markers we first performed cohort-level EWAS of EA that we subsequently meta-analysed (Supplementary Note 2). As is standard, the EWAS was performed as a set of linear regressions in each cohort, one methylation marker at a time, with the methylation beta value (0–1) as the dependent variable. The key independent variable was EA. We estimated two regression models that differ in the set of covariates included. In the basic model, the covariates were age, sex, imputed or measured white blood cell counts, technical covariates from the methylation array and four genetic principal components to account for population stratification. In the adjusted model, we additionally controlled for BMI (kg/m2), smoker status (three categories: current, previous or never smoker), an interaction term between age and sex and a quadratic term for age. As BMI and smoking are correlated with EA38, 39 and known to be associated with methylation,13, 17 the basic model may identify associations with EA that are actually due to BMI or smoking. Although the adjusted model reduces that risk, it may also reduce power to identify true associations with EA (by controlling for factors that are correlated with EA). Although we present the results for both models, we focus on the adjusted model because it is more conservative. Details of cohort-specific covariates are presented in Supplementary Table S1.4.
EWAS QC and meta-analysis
Each participating cohort uploaded EWAS summary statistics to a central secure server for QC and meta-analysis. The number of CpG probes filtered at each step of the QC is presented in Supplementary Table S1.5. We removed: probes with missing P-value, s.e. or coefficient estimate; probes with a call rate <95%; probes with a combined sample size <1000; probes not available in the probe-annotation reference by Price et al.;40 CpH probes (H=A/C/T); probes on the sex chromosomes; and cross-reactive probes highlighted in a recent paper by Chen et al.41 We performed a sample-size-weighted meta-analysis of the cleaned results using METAL.42 We used single genomic control, as is common in genome-wide association studies (GWAS),43 to stringently correct the meta-analysis P-values for possible unaccounted for population stratification.44 Probes with a P-value<1 × 10−7, a commonly used threshold in EWAS1 that we prespecified in the analysis plan, were considered epigenome-wide significant associations.
Epigenetic clock analyses
To construct our epigenetic clock variables (Supplementary Note 3), the cohort-level raw beta-value data were entered into the online Horvath calculator,23 as per our preregistered analysis plan. The ‘normalize data’ and ‘advanced analysis for Blood Data’ options were selected. The following variables were selected from the calculator’s output for subsequent analysis:
-
Clock 1. Horvath age acceleration residuals, which are the residuals from the regression of chronological age on Horvath age.
-
Clock 2. White blood cell count-adjusted Horvath age acceleration, which is the residual from Clock 1 after additional covariate adjustment for imputed white blood cell counts.
-
Clock 3. White blood cell count-adjusted Hannum age acceleration, which is the same as Clock 2 but with the Hannum age prediction in place of the Horvath prediction.
-
Clock 4. Cell count-enriched Hannum age acceleration, which is the basic Hannum predictor plus a weighted average of aging-associated cell counts. This index has been found to have the strongest association with mortality.45
These Clock measures are annotated in the Horvath software as follows: ‘AgeAccelerationResidual’, ‘AAHOAdjCellCounts’, ‘AAHAAdjCellCounts’, and ‘BioAge4HAAdjAge’. We analyzed two regression models, both with EA as the dependent variable and a clock variable as an independent variable. In the basic age acceleration model, we control for sex, and in the adjusted age acceleration model, we additionally control for BMI and smoker status (current, previous or never smoker). In total, in each adult cohort, we estimated eight regressions: each of the two models with each of the four clock variables as an independent variable. For each of the eight regressions, we performed a sample-size-weighted meta-analysis of the cohort-level results.
Polygenic predictions with polygenic methylation score
We performed a prediction analysis with polygenic methylation scores (PGMSs), analogous to polygenic score prediction in the GWAS literature (Supplementary Note 6). We tested the predictive power in three independent adult cohort studies: Lothian Birth Cohort 1936 (LBC1936, n=918), RS-BIOS (Rotterdam Study—BIOS, n=671), and RS3 (Rotterdam Study 3, n=728). We re-ran the EWAS meta-analysis for each prediction cohort to obtain the weights for the PGMS, while holding out the prediction cohort to avoid overfitting. We constructed the PGMS for each individual as a weighed sum of the individual’s methylation markers’ beta values and the EWAS effect-sizes, using the Z-statistics from the EWAS as weights. (The Z-statistics were used instead of the EWAS coefficients because CpG methylation is the dependent variable in the EWAS regression.). We constructed PGMSs using two different thresholds for probe inclusion, P<1 × 10−5 and P<1 × 10−7, with weights from the basic and adjusted EWAS models, for a total of four PGMSs in each prediction cohort.
To shed light on the direction of causation of epigenetic associations, we used a fourth prediction cohort study, a sample of children in the ALSPAC ARIES cohort.47 We constructed the PGMS using the same approach as described above, in this case using data from cord-blood-based DNA methylation at birth. The outcome variables in this cohort were average educational achievement test scores (Key Stages 1–448) from age 7 up to age 16 years.
To examine the relationship between epigenetic and genetic associations, we also constructed a single-nucleotide polymorphism polygenic score (SNP PGS) for EA. We used SNP genotype data available in the three adult prediction cohort studies (LBC1936, RS-BIOS and RS3). We constructed the SNP PGS in each cohort as a weighted sum of the individual genotypes from all available genotyped SNPs, with GWAS meta-analysis coefficients as weights. We obtained the coefficients by re-running the largest GWAS meta-analysis to date of EA35 after excluding our prediction cohorts (LBC1936, RS-BIOS and RS3).
We evaluated the predictive power of the PGMS by examining the incremental coefficient of determination (incremental R2) for predicting EA (or test scores in ALSPAC ARIES). The incremental R2 is the difference in R2 between the regression model with only covariates and the same regression model that additionally includes the PGMS as a predictor. The covariate-only models in the LBC1936, RS-BIOS and RS3 cohorts controlled for age, sex and the SNP PGS. In the ALSPAC ARIES cohort, we controlled for age at assessment and sex. In the ALSPAC ARIES cohort, when we investigate maternal smoking as a potential confound for our EA associations, we add maternal smoking to the set of covariates. We finally restricted the ALSPAC ARIES cohort to children with non-smoking mothers. To investigate a possible interaction effect between the PGMS and SNP PGS, we re-estimated the regression model after adding an interaction term between the PGMS and the SNP PGS, and the incremental R2 was calculated as the difference in R2 relative to the model that included the PGMS and the SNP PGS as additive main effects.
Results
Descriptive statistics
Summary statistics from the 27 independent cohort studies from the 15 contributing cohorts are shown in Supplementary Table S1.1. The mean age at reporting ranges from 26.6 to 79.1 years, and the sample size ranges from 48 to 1658, with a mean of 399 individuals per cohort. The mean cohort EA ranges from 8.6 to 18.3 years of education, and the sample-size-weighted mean is 13.6 (s.d.=3.62). The meta-analysis sample is 54.1% female.
Epigenome-wide association study
EWAS quality control
The QC filtering is reported in Supplementary Table S1.5. We inspected the quantile–quantile (QQ) plot of the filtered EWAS results from each contributing cohort as part of the QC procedure before meta-analysis. The genomic inflation factor (λGC), defined as the ratio of the median of the empirically observed chi-square test statistics to the expected median under the null, had a mean across the cohorts of 1.02 for the adjusted model (s.d.=0.18). We report the cohort-level genomic inflation factor after probe filtering in Supplementary Table S1.5. The variation in λGC across cohorts was comparable to that from EWAS performed in cohorts of similar sample size.12 We applied genomic control at the cohort level, which is a conservative method of controlling for residual population stratification that may remain even despite the regression controls for principal components.44 The meta-analysis λGC was 1.19 for the basic model and 1.06 for the adjusted model.
EWAS results
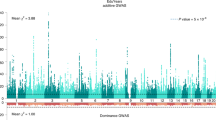
Figure 1 shows the Manhattan plot for the meta-analysis results of the adjusted model. The Manhattan plot for the basic model is reported in Supplementary Note, together with the QQ plots for the basic and adjusted model. In the basic model, there were 37 CpG probes associated with EA at our preregistered epigenome-wide P-value threshold (P<1 × 10−7); these results are reported in Supplementary Table S1.6a. In the adjusted model, there were nine associated probes, listed in Table 1 (with additional details in Supplementary Table S1.7a), all of which were also associated in the basic model. We hereafter refer to the adjusted model’s nine associated probes as the ‘lead probes’. In Supplementary Note 2.4.2, we present the association results with false discovery rate <0.05, but as this threshold was not prespecified in the analysis plan, we do not present these results as main findings.

Manhattan plot of the adjusted epigenome-wide association study (EWAS) model. The figure displays the Manhattan plot of the meta-analysis of the adjusted EWAS model (the Manhattan plot of the basic model is reported in Supplementary Note). The x axis is the chromosomal position, and the y axis is the significance on a −log10 scale. The dashed lines mark the threshold for epigenome-wide significance (P=1 × 10−7) and for suggestive significance (P=1 × 10−5). Each epigenome-wide associated probe is marked with a red × and the symbol of the closest gene based on physical position.
To investigate how the EWAS results look at a regional level, we analyzed the distribution of the EWAS associations across the genome by performing enrichment tests for methylation density regions40 (the so-called ‘HIL’ categories; Supplementary Note 5.2). We found that the number of probes with P<1 × 10−7 is more or less proportional to the total number of probes in every region and that there is enrichment for association in all four methylation density categories: high-density CpG islands (HC), intermediate-density CpG islands (IC), intermediate-density CpG islands bordering HCs (ICshore), and non-islands (LC).
The effect sizes of the associations for the nine lead probes are shown in Table 1. The coefficients of determination (R2s) range from 0.3% to 0.7%. To put these effect sizes in perspective, Figure 2 and Supplementary Table S1.8 compare the R2s for the top 50 probes in our adjusted model with the top 50 probes from recent large-scale EWAS of smoking,13 maternal smoking,12 alcohol consumption15 and BMI,17 as well as the top 50 GWAS SNP associations with EA.35 The EA EWAS associations of our study are an order of magnitude larger than the largest EA SNP effect sizes. However, our EWAS associations are small in magnitude relative to the EWAS associations reported for more biologically proximal environmental factors. BMI is the most similar to EA, with R2s of associated probes approximately 20–50% larger than those for EA. Relative to the largest R2 for an EA-associated probe, the largest effect for probes associated with smoking and maternal smoking are greater by factors of roughly 3 and 17, respectively.

Epigenome-wide association study (EWAS) effect sizes (in terms of variance explained) across traits and with genome-wide association study (GWAS). The figure displays the effect size estimates in terms of R2, in descending order, for the 50 top probes of the adjusted EWAS model. For comparison, we present the 50 top probes from recent EWAS on alcohol consumption (n=9643, Liu et al., 2016), body mass index (BMI; n=7798, Mendelson et al., 2017), smoking (n=9389, Joehanes et al., 2016) and maternal smoking (n=6685, Joubert et al.12). For comparison with GWAS effect sizes, we contrast the EWAS probes with the effect sizes of the 50 top approximately independent single-nucleotide polymorphisms (SNPs) from a recent GWAS on educational attainment (EA; n=405 073, Okbay et al., 2016). Panels (a and b) display the same results but with a different scaling of the y axis in order for the smaller effect sizes to be visible.
Lookup of lead probes in the published EWAS of smoking
As our smoker-status control variable is coarse and discrete (current, former or never smoker), we were concerned that the adjusted EWAS model might not have adequately controlled for exposure to smoking (that is, amount and duration of smoking and exposure to second-hand smoke). Therefore, we performed a lookup of our lead probes in the published EWAS on smoking (Supplementary Note 4 and Supplementary Table S1.10). We found that all nine lead probes have previously been associated with smoking. The results of this lookup motivated our analysis of the never-smoker subsample, discussed next.
Robustness of EWAS results in the never-smoker subsample
To minimize the possible confounding effect of smoking on the association between EA and CpG methylation, we conducted a set of analyses that we did not anticipate when we preregistered our analysis plan. Specifically, we went back to the cohorts and asked them to re-conduct their EWAS, this time restricting the analysis to individuals who self-reported to be never smokers. After following the same QC steps as above, we performed a new meta-analysis of these results (n=5175).
In this subsample, the effect-size estimates were smaller by at least 60% for 7 of the 9 lead probes (see Table 1 and Figure 3a), whereas two probes (cg12803068 and cg22132788) had similar effect-size estimates as in the full sample (statistically distinguishable from 0 with P=1.48 × 10−4 and P=4.35 × 10−4, respectively). These two probes, however—both in proximity to the gene MYO1G—have been found to be associated with maternal smoking during pregnancy, and the effects on the methylation of this gene are persistent when measured at age 17 years in the offspring.12, 49 This influence has been shown to continue through to middle age.50 We cannot distinguish between the hypothesis that these probes have some true association with EA and the hypothesis that their apparent association with EA is entirely driven by more maternal smoking during pregnancy among lower EA individuals. We also cannot rule out that the probes’ association with EA is driven by second-hand smoke exposure, which could also be correlated with EA.

Comparison of educational attainment (EA) epigenome-wide association study (EWAS) effect sizes with the effect sizes in the never-smoker subsample and in smoking EWAS results. Panel (a) displays the effect-size estimates in terms of R2 for the nine lead probes, in descending order, and the lead probe’s corresponding effect size when re-estimated in the subsample of never smokers. Panel (b) displays the same information for the probes of the adjusted model with P<1 × 10−5 (including the nine lead probes), as well as the same probes’ effect-size estimates from two recent EWAS of smoking (n=9389, Joehanes et al.13), and maternal smoking (n=6685, Joubert et al.12). The smoking and maternal smoking estimates are only publicly available for probes associated at false discovery rate <0.05 in the respective EWAS.
To assess how widely such confounding may affect the EA results, in Figure 3b we compare the effect sizes of all the probes associated with EA at P<1 × 10−5 in the adjusted EWAS model to the effect sizes found for the same probes in EWAS meta-analyses of smoking13 and maternal smoking12 (see also Supplementary Table S.1.11). Many of the EA-associated probes are also associated with smoking or maternal smoking, strongly suggesting that residual smoking exposure (that is, the misclassification of amount and duration of smoking and second-hand smoke that is not captured by the smoking covariate) and maternal smoking remain potential confounding factors for the probe associations with EA, even in the subsample of individuals who are self-reported never smokers.
Epigenetic clock associations with EA
Two cohorts, FINRISK and MCCS, did not contribute to the epigenetic clock analyses. Therefore, the sample sizes for these analyses were smaller than for the EWAS meta-analysis: 8173 for the basic age acceleration model and 7691 for the adjusted age acceleration model (the difference being due to missing covariates for some individuals). The effect-size estimates are presented in Figure 4 and Supplementary Table S1.9. There was no evidence for an association between EA and Clocks 1, 2 or 3, but the association between EA and Clock 4 was strong (P=3.51 × 10−6 and P=4.51 × 10−4 in the basic and adjusted age acceleration models, respectively). The point estimates were small, however: using Clock 4, each year of EA was associated with a 0.071-year (that is, 26-day) reduction in age acceleration in the basic model and a 0.055-year (that is, 20-day) reduction in the adjusted model. Overall then, higher EA was associated with slightly younger biological age when compared with chronological age. We note that the epigenetic clock that was found to be associated with EA, Clock 4, has previously been found to be the most predictive epigenetic clock measure of mortality,45 and an independent study also found a negative association between Clock 4 and EA46

Effect-size estimates (in days) of the epigenetic clock analyses with 95% confidence intervals. Panel (a) displays the effect-size estimates from the basic age acceleration model, and panel (b) displays the effect-size estimates from the adjusted age acceleration model. The effect size is denoted in days of age acceleration per year of educational attainment, and error bars represent 95% confidence intervals.
Prediction using PGMSs
The incremental R2s from the prediction of EA with PGMSs in our adult prediction cohort studies, the LBC1936, RS-BIOS and RS3, are reported in Supplementary Table S1.13a and Figure 5. Across the four PGMSs constructed with weights from the basic and adjusted model, and with the two probe-inclusion thresholds (P<1 × 10−5 and P<1 × 10−7), the incremental R2s ranged from 1.4% to 2.0% (P⩽3.28 × 10−8). There was also weak evidence for an interaction between the PGMS and the SNP PGS in predicting EA, with the R2s for the interaction term ranging from 0.1% to 0.3% (P-values ranged from 0.01 to 0.12).

Methylation score prediction of educational attainment in independent holdout samples. Panel (a) displays the prediction in all individuals, and panel (b) displays the prediction in the subsample of never smokers. Four methylation scores were constructed using coefficient estimates from the basic model versus adjusted model, crossed with a P-value threshold of 1 × 10−5 and 1 × 10−7. The sample sizes of the LBC1936, the RS3 and the RS-BIOS cohorts are 918, 728 and 671 individuals, respectively. We performed sample-size-weighted meta-analysis across the cohorts for each of the four methylation score prediction analyses. From left to right, the respective P-values testing the null hypothesis of zero predictive power are 4.42 × 10−11, 7.76 × 10−11, 2.02 × 10−11 and 3.28 × 10−8 for the full sample and 0.0183, 0.0898, 0.0051 and 0.1818 for the never smokers, respectively. The full prediction results are presented in Supplementary Tables S1.9a and b.
In the subsample of never smokers, the PGMSs (constructed with weights derived from the full EWAS sample), the PGMS is far less predictive, with incremental R2s ranging from 0.3% to 0.9% (Figure 5 and Supplementary Table S1.13b). The two PGMSs constructed from probes with P<1 × 10−5 in the EWAS were associated with EA at P<0.05, while the two PGMSs constructed only from the lead probes with P<1 × 10−7 were not (P>0.05). No interaction effect was found between the PGMS and the SNP PGS in the never-smoker subsample.
To further investigate confounding by smoking in the prediction analysis, we examined the correlations between our PGMSs constructed from the lead probes (that is, those associated with EA at significance threshold P<1 × 10−7) in either our basic or adjusted model and a PGMS for smoking (see Supplementary Note 6.2.2 for details). For the smoking PGMS, we use the 187 probes that were identified at epigenome-wide significance (P<1 × 10−7) and then successfully replicated in a recent EWAS of smoking.51 We examine the PGMS correlations in our full prediction samples, not restricted to never smokers. For the EA PGMS from our basic model, we find a correlation with the smoking PGMS of −0.96 in RS3, −0.94 in RS-BIOS and −0.93 in LBC1936. For the EA PGMS from our adjusted model, the correlations are −0.90, −0.89 and −0.91, respectively. In all cases, the nearly perfect correlation between the smoking and EA methylation scores strongly suggests that smoking status confounds the EWAS associations with EA.
Turning to the child sample in the ALSPAC ARIES cohort,47, 49 we examined whether a PGMS constructed from methylation assessed in cord blood samples at birth was predictive of four prospective measures of educational achievement test scores (Key Stage 1–448), collected between ages 7 and 16 years (Supplementary Note 6.1.1). The results are reported in Supplementary Table S1.13c. The largest incremental R2 was 0.73% (P=0.0094), and it was attained in the model predicting school performance at age 14–16 years (that is, the Key Stage 4 test scores). However, once maternal smoking status was added as a control variable, the predictive power of the PGMS became essentially zero (incremental R2=0.05%, P=0.234). This suggests that the confounding effects of maternal smoking strongly influenced the predictive power of the PGMS for EA. We draw two conclusions from these results from the child sample. First, they reinforce the concern that maternal smoking was a major confound for any probe associations with EA. Second, they suggest that any true methylation–EA associations were unlikely to be driven by a causal effect of methylation on EA.
Overlap between EWAS probes and published GWAS associations
To supplement our polygenic score analyses of the overlap between epigenetic and genetic associations, we next investigated whether our lead probes are located at loci that contain SNPs previously identified in GWAS of EA and smoking (Supplementary Note 5). Considering jointly the 141 approximately independent EWAS probes with P<1 × 10−4, we did not find evidence of enrichment for either EA-linked SNPs (P=0.206) or smoking-linked SNPs (P=0.504). Considering the probes individually, one probe (cg17939805) was found to be in the same genomic region as a SNP (rs9956387) associated with EA (with a genomic distance of 607 bp), whereas no probes were close to SNPs previously identified as linked to smoking.
Correlation of EWAS results with tissue-specific methylation
To answer the question of whether our EWAS associations are correlated with any tissue-specific DNA methylation, we utilized the tissue-specific methylation data made available by the Epigenomic Roadmap Consortium.52 Their data were used to calculate tissue-specific deviations from average cross-tissue methylation at the loci corresponding to the EWAS CpG probes associated with EA at a P-value<1 × 10−4 (Supplementary Note 7). We examined the correlation between these tissue-specific deviations and the EWAS association test statistics (Z-statistics) of the probes, using the results from the adjusted EWAS model. We report the results in Figure 6 and in Supplementary Table S1.14. The strongest correlations were found for primary haematopoietic stem cells granulocyte colony-stimulating factor-mobilized female and IMR90 fetal lung fibroblast. Intermediate strength correlations were found across multiple, seemingly unrelated tissues, while no correlations of relevant magnitudes were found with the brain tissues available in the Roadmap. We interpret the lack of correlation with tissues plausibly related to EA (such as brain tissues) as supporting the conclusion that the EWAS results are driven by confounding factors rather than by a true association with EA.

Correlations between tissue-specific methylation and the epigenome-wide association study association results (adjusted model). Panel (a) displays the correlation estimates based on the whole-genome bisulfite sequencing methylation measurement, and panel (b) displays results based on the mCRF methylation measurement. (The mCRF measurement combines sequencing data from the MeDIP-seq and MRE-seq methods.) The method is described in Supplementary Note 7. Correlations that are significant after Bonferroni correction are marked with two asterisks (**), and marginal significance (P<0.05) is marked with one asterisk (*). The tissue-specific methylation data are from the Roadmap Epigenomics Consortium, and we used their categorization and color code for simplicity of comparison.52
Pathway analysis with gene-expression data
Using the GTEx53 expression data and the webtool ‘functional mapping and annotation of genetic associations’ (FUMA)54 we performed a pathway analysis. The analysis used the GTEx gene-expression levels to cluster the 29 genes physically closest to the EA-associated (at P<1 × 10−5) CpG probes of the adjusted model (Supplementary Note 8). The results of the expression analysis are displayed in Supplementary Figure 4. We find that the genes closest to the EA-associated probes are expressed across multiple tissues that have no clear relationship to EA (such as blood tissues, among many other); for further discussion, see Supplementary Note. Overall, these results are consistent with the hypothesis that the EWAS results are driven by confounding factors.
Discussion
We believe this study provides one of the first large-scale investigations in humans of epigenetic changes linked to a biologically distal environmental factor. In our EWAS meta-analysis—one of the largest EWAS conducted to date—we found nine CpG probes associated with EA. Each of these probes explains 0.3–0.7% of the variance in EA—effect sizes somewhat smaller than the largest EWAS effects that have been observed for BMI and many times smaller than those observed for alcohol consumption, smoking and especially maternal smoking during pregnancy. When we restrict our analysis to the subsample of never smokers, the effect sizes of seven out of the nine lead probes are substantially attenuated. Moreover, the other two lead probes have been found in previous work to be strongly associated with maternal smoking during pregnancy.12 More generally, comparing our own results to those from previous EWAS highlights a variety of factors correlated with EA, including not only maternal smoking but also alcohol consumption and BMI, as potentially major confounding factors for the EA associations we detect. We also cannot rule out that other factors correlated with EA, such as exposure to second-hand smoke, could confound the EA associations. This should be taken into account in future endeavours of associating methylation with biologically distal factors that are known to correlate with environmental factors that have a fairly direct biological impact, such as smoking.
Convincingly establishing a causal effect of EA would require analyzing a sample with quasi-random variation in EA, such as a sample in which some individuals were educated after an increase in the number of years of compulsory schooling and other individuals were educated before the law change.55 We are not aware of large EWAS samples with quasi-random variation at present, but we anticipate that such samples will become available as methylation becomes more widely measured.
Although the EWAS we report here is among the largest conducted to date, our sample size of 10 767 individuals is only large enough to identify nine probes associated with EA at the conventional epigenome-wide significance threshold. Subsequent EWAS conducted in larger samples that have sufficient statistical power to identify a much larger number of EA-associated probes will enable more extensive investigations of overlap with probes associated with other phenotypes than were possible from our results, as well as analyses of the biological functions of the probes. Besides limited statistical power, other limitations of our study, common to EWAS research designs, are that we study methylation cross-sectionally and not longitudinally and that we only investigate CpG methylation and not other types of epigenetic modifications. Also, our study focuses on single CpG sites; future studies could consider additional analytical approaches to assess regions of differential methylation (for example, genes). Once suitable methods have been developed, it would also be of interest to estimate the overall proportion of variance in EA that can be attributed to individual differences in DNA methylation patterns.
Conclusion
One plausible hypothesis is that environmental influences on the epigenome—even those due to everyday, social environmental factors—are pervasive and profound.3 According to the logic of this view, a major life experience that occurs over many years, such as EA, should leave a powerful imprint on the epigenome. Motivated by this view and by the evidence of large EWAS effects in studies of lifestyle factors, when we embarked on this project we entertained the hypothesis that we might find large associations between EA and methylation. We also entertained the alternative hypothesis that EA, because it is so biologically distal, may exhibit much weaker associations with methylation.
Although our results do not allow us to distinguish how much of the effects we find are due to true associations with EA and how much are due to confounding factors, they strongly suggest that the effect sizes we estimate are an upper bound on the effect sizes of any true methylation associations with EA. These upper-bound effect sizes are far smaller than associations with more biologically proximal environmental factors that have been studied. If our results can be generalized beyond EA to other biologically distal environmental factors, then they cast doubt on the hypothesis that such factors have large effects on the epigenome.
References
Rakyan VK, Down TA, Balding DJ, Beck S . Epigenome-wide association studies for common human diseases. Nat Rev Genet 2012; 12: 529–541.
Veldhoven KV, Polidoro S, Baglietto L, Severi G, Sacerdote C, Panico S et al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin Epigenetics 2015; 7: 67–79.
Champagne FA, Curley JP Epigenetic influence of the social environment. In: Petronis A, Mill J (eds). Brain, Behavior, and Epigenetics. Springer-Verlag Berlin Heidelberg: Heidelberg, Germany, 2011, pp 23–40.
Beach SRH, Lei M-K, Brody GH, Kim S, Barton AW, Dogan MV et al. Parenting, socioeconomic status risk, and later young adult health: exploration of indirect effects via DNA methylation. Child Dev 2016; 87: 111–121.
Borghol N, Suderman S, McArdle W, Racine A, Hallet M, Pembrey M et al. Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol 2012; 41: 62–74.
Cunliffe VT . The epigenetic impacts of social stress: how does social adversity become biologically embedded? Epigenomics 2016; 8: 1653–1669.
Jones-Mason K, Allen IE, Bush N, Hamilton S . Epigenetic marks as the link between environment and development: examination of the associations between attachment, socioeconomic status, and methylation of the SLC6A4 gene. Brain Behav 2016; 6: 1–18.
Stringhini S, Polidoro S, Sacerdote C, Kelly RS, Veldhoven K, Agnoli C et al. Life-course socioeconomic status and DNA methylation of genes regulating inflammation. Int J Epidemiol 2015; 44: 1320–1330.
Szyf M . DNA methylation, the early-life social environment and behavioral disorders. J Neurodev Disord 2011; 3: 238–249.
Szyf M, McGowan P, Meany MJ . The social environment and the epigenome. Environ Mol Mutagen 2008; 49: 46–60.
Gao X, Jia M, Zhang Y, Breitling LP, Brenner H . DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics 2015; 7: 113–123.
Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet 2016; 98: 680–696.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet 2016; 9: 436–447.
Ungerer M, Knezovich J, Ramsay M . In utero alcohol exposure, epigenetic changes, and their consequences. Alcohol Res 2013; 35: 37–46.
Liu C, Marioni RE, Hedman ÅK, Pfeiffer L, Tsai P-C, Reynolds LM et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry 2016 e-pub ahead of print 15 November 2016; doi: 10.1038/mp.2016.192.
Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson MM, Zhou Y-H et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet 2015; 24: 4464–4479.
Mendelson MM, Marioni RE, Joehanes R, Liu C, Hedman ÅK, Aslibekyan S et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med 2017; 14: 1–30.
Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol 2016; 17: 1–15.
Ioannidis JPA . Why most published research findings are false. PLoS Med 2005; 2: 696–701.
Benjamin DJ, Cesarini D, Chabris CF, Glaeser EL, Laibson DI, Gudnason V et al. The promises and pitfalls of genoeconomics. Annu Rev Econom 2012; 4: 627–662.
Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Lifang H et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11: 587–596.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49: 359–367.
Horvath S . DNA methylation age of human tissues and cell types. Genome Biol 2013; 14: 1–19.
Rönn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet 2015; 24: 3792–3813.
Houtepen LC, Vinkers CH, Carrillo-Roa T, Hiemstra M, Lier PA, Meeus W et al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat Commun 2016; 7: 1–10.
Masemola ML, Merwe L, Lombard Z, Viljoen D, Ramsay M . Reduced DNA methylation at the PEG3 DMR and KvDMR1 loci in children exposed to alcohol in utero: a South African Fetal Alcohol Syndrome cohort study. Front Genet 2015; 5: 1–12.
Barrow TM, Michels KB . Epigenetic epidemiology of cancer. Biochem Biophys Res Commun 2014; 455: 70–83.
Marioni RE, Harris SE, Shah S, McRae AF, Zglinicki T, Martin-Ruiz C et al. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol 2016; 45: 424–432.
Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol 2015; 44: 1388–1396.
Levine ME, Lu AT, Bennett DA, Horvath S . Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015; 7: 1198–1211.
Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015; 14: 491–495.
Zannas AS, Arloth J, Carrillo-Roa T, Iurato S, Röh S, Ressler KJ et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol 2015; 16: 266–278.
Simons RL, Lei MK, Beach SRH, Philibert RA, Cutrona CE, Gibbons FX et al. Economic hardship and biological weathering: the epigenetics of aging in a U.S. sample of black women. Soc Sci Med 2016; 150: 192–200.
Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 2013; 340: 1467–1471.
Okbay A, Beauchamp JP, Fontana MA, Lee JJ, Pers TH, Rietveld CA et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 2016; 533: 539–542.
Triche TJ, Weisenberger DJ, Berg D, Laird PW, Siegmund KD . Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res 2013; 41: 1–11.
Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 2014; 15: 503–520.
Johnson W, Kyvik KO, Mortensen EL, Batty GD, Deary IJ . Does education confer a culture of healthy behavior? Smoking and drinking patterns in Danish twins. Am J Epidemiol 2011; 173: 55–63.
Hermann S, Rohrmann S, Linseisen J, May AM, Kunst A, Besson H et al. The association of education with body mass index and waist circumference in the EPIC-PANACEA study. BMC Public Health 2011; 11: 169–181.
Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 2013; 6: 4–19.
Chen Y-A, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8: 203–209.
Willer CJ, Li Y, Abecasis GR . METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–2191.
Devlin B, Roeder K . Genomic control for association studies. Biometrics 1999; 55: 997–1004.
Iterson M, Zwet E, the BIOS Consortium, Heijmans BT . Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol 2017; 18: 1–13.
Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai P-C et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging 2016; 8: 1844–1865.
Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Ageing 2017; 9: 419–437.
Relton CL, Gaunt T, McArdle W, Ho K, Duggirala A, Shihab H et al. Data resource profile: accessible resource for integrated epigenomic studies (ARIES). Int J Epidemiol 2015; 44: 1181–1190.
GOV.UK. National Curriculum [Internet]. 2016 [cited 14 January 2017]. Available from https://www.gov.uk/national-curriculum.
Richmond RC, Simpkin AJ, Woodward G, Gaunt TR, Lyttleton O, McArdle WL et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet 2015; 24: 2201–2217.
Richmond RC, Suderman M, Langdon R, Relton CL, Davey Smith G . DNA methylation as a marker for prenatal smoke exposure in adults. bioRxiv 2017 e-pub ahead of print 28 March 2017; doi: https://doi.org/10.1101/121558.
Zeilinger S, Kühnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013; 8: e63812.
Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Erst J, Bilenky M, Yen A et al. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518: 317–330.
The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013; 45: 580–585.
Watanabe K, Taskesen E, Bochoven A, Posthuma D . FUMA: functional mapping and annotation of genetic associations. bioRxiv 2017 e-pub ahead of print 20 February 2017; doi: https://doi.org/10.1101/110023.
Lager ACJ, Torssander J . Causal effect of education on mortality in a quasi-experiment on 1.2 million Swedes. Proc Natl Acad Sci 2012; 109: 8461–8466.
Acknowledgements
This research was carried out under the auspices of the Social Science Genetic Association Consortium (SSGAC). The SSGAC seeks to facilitate studies that investigate the influence of genes on human behavior, well-being and social-scientific outcomes using large genome-wide association study meta-analyses. The SSGAC also provides opportunities for replication and promotes the collection of accurately measured, harmonized phenotypes across cohorts. The SSGAC operates as a working group within the CHARGE consortium. A full list of acknowledgments is provided in Supplementary Note. Upon publication, results can be downloaded from the SSGAC website (http://www.thessgac.org/). Data for our analyses come from many studies and organizations, some of which are subject to a MTA, and are listed in the Supplementary Note. We thank Aysu Okbay for conducting the meta-analyses for the SNP polygenic scores. The data were accessed under Section 4 of the Data Sharing Agreement of the SSGAC.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Karlsson Linnér, R., Marioni, R., Rietveld, C. et al. An epigenome-wide association study meta-analysis of educational attainment. Mol Psychiatry 22, 1680–1690 (2017). https://doi.org/10.1038/mp.2017.210
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.210
This article is cited by
-
Trans-ancestry epigenome-wide association meta-analysis of DNA methylation with lifetime cannabis use
Molecular Psychiatry (2024)
-
Maternal educational attainment in pregnancy and epigenome-wide DNA methylation changes in the offspring from birth until adolescence
Molecular Psychiatry (2024)
-
Identification of novel hypermethylated or hypomethylated CpG sites and genes associated with anthracycline-induced cardiomyopathy
Scientific Reports (2023)
-
A comparison of the genes and genesets identified by GWAS and EWAS of fifteen complex traits
Nature Communications (2022)
-
Epigenome-wide meta-analysis identifies DNA methylation biomarkers associated with diabetic kidney disease
Nature Communications (2022)
