
Abstract
Maternal educational attainment (MEA) shapes offspring health through multiple potential pathways. Differential DNA methylation may provide a mechanistic understanding of these long-term associations. We aimed to quantify the associations of MEA with offspring DNA methylation levels at birth, in childhood and in adolescence. Using 37 studies from high-income countries, we performed meta-analysis of epigenome-wide association studies (EWAS) to quantify the associations of completed years of MEA at the time of pregnancy with offspring DNA methylation levels at birth (n = 9 881), in childhood (n = 2 017), and adolescence (n = 2 740), adjusting for relevant covariates. MEA was found to be associated with DNA methylation at 473 cytosine-phosphate-guanine sites at birth, one in childhood, and four in adolescence. We observed enrichment for findings from previous EWAS on maternal folate, vitamin-B12 concentrations, maternal smoking, and pre-pregnancy BMI. The associations were directionally consistent with MEA being inversely associated with behaviours including smoking and BMI. Our findings form a bridge between socio-economic factors and biology and highlight potential pathways underlying effects of maternal education. The results broaden our understanding of bio-social associations linked to differential DNA methylation in multiple early stages of life. The data generated also offers an important resource to help a more precise understanding of the social determinants of health.
Similar content being viewed by others


Introduction
Maternal educational attainment (MEA) is a multidimensional construct that influences child health and wellbeing via myriad social and biological pathways [1]. Among the core components of socio-economic position (SEP) i.e. employment, income, and education, MEA shows the strongest association with child neuro-cognitive development. It determines access to important resources, such as financial security, family circumstances, and material resources, that affect child birthweight, growth and development and cardio-metabolic health in later life [2].
MEA has been shown to influence other relevant intrauterine exposures such as nutrition, maternal smoking, body mass index etc. that are related to child health outcomes. Part of the downstream impact of intrauterine exposures on offspring health has been found to be through altered DNA methylation. Despite widespread recognition of social factors in health, prospective evidence for underlying mechanisms of this ‘biological embedding’ from an early time point is limited, and causal mechanisms are unknown. Recent research has revealed epigenetic variation associated with SEP and discovered that, when compared to other markers of SEP [3] education (either one’s own or one’s mother’s) has the largest influence. Similarly, only maternal education was related with four cytosine-phosphate-guanine (CpG) sites at birth and twenty in adolescence, according to a longitudinal analysis of 974 participants from the ALSPAC birth cohort (United Kingdom) [4]. With respect to own education, Linner et al. in a study including 10 767 participants from 27 cohorts within Social Science Genetics Association Consortium (SSGAC) identified nine CpGs related to educational attainment in adults aged 26.6–79.1 years, overlapping with findings from previous studies on adult smoking and maternal smoking during pregnancy [5].
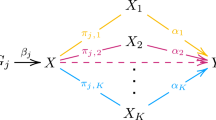
A low level of maternal education is not a sufficient cause of offspring health per se, but it may mediate a vulnerability increasing the risk to be exposed to other prenatal exposures with direct effects on DNA methylation (Fig. 1). We aimed to quantify the associations of MEA with DNA methylation levels at birth, in childhood and in adolescence. Here we present meta-analyses of multiple EWASs in 37 studies from high income countries, with sample size of up-to 9881 individuals. We explored (i) if findings are enriched with those from EWASs of intrauterine exposures with clear impacts on offspring methylation, thereby indicating that MEA may serve as a proxy for better health behaviours; and (ii) association of implicated sites with gene expression in cells and tissues.

Gray dotted arrows denote plausible measures that may be linked with MEA and DNA methylation.
Methods
Participating cohorts
The study included 37 studies from high income countries in Europe, the USA and Australia within the Pregnancy And Childhood Epigenetics (PACE) Consortium [6]. The total sample across the three time points included 14,638 individuals comprising 96.3% European, 1.8% Hispanic, and 1.7% African ethnicity. Ethnicity was self-reported unless stated otherwise in the cohort specific methods (Supplementary File). DNA methylation was measured in offspring at three time points: birth (27 studies, n = 9 881), childhood (6 studies, n = 2 017), and adolescence (4 studies, n = 2 740). Participants had complete information on MEA, DNA methylation in cord blood or peripheral blood, and the covariates described below (complete case analysis). We excluded all twins and in case of non-twin siblings, one sibling was excluded by selecting based on completeness of data or, if equal, randomly.
Written informed consent was obtained for all participants, and studies were approved by the local ethics boards in accordance with the principles of the Declaration of Helsinki. Supplementary methods provide cohort-specific detailed information, and their ethics approval statements (Supplementary File).
Maternal education measures
MEA at the time of pregnancy was defined in accordance with the International Standard Classification of Education (ISCED) 1997 classification (UNESCO) [7] and was harmonized across the cohorts. MEA was categorized into seven categories (coded 0 to 6) of educational attainment, which was then translated into years of schooling equivalents (0 to 22 years of schooling) as: Level 0 = 1 year, Level 1 = 7 years, Level 2 = 10 years, Level 3 = 13 years, Level 4 = 15 years, Level 5 = 19 years and Level 6 = 22 years of schooling (Supplementary Table 1).
DNA methylation measurement
All cohorts extracted DNA from cord blood and/or peripheral blood samples. Samples were processed with the Infinium HumanMethylation450 or EPIC BeadChip assays [8]. Quality control and normalization were performed independently by the individual cohorts (Supplementary File). Untransformed beta-values were used as the outcome measure (DNA methylation beta-values 0-1). Methylation value outliers were excluded using the Tukey method: values < (25th percentile- 3IQR) and values > (75th percentile +3IQR) were removed [9]. CpGs located on the sex chromosomes were also removed.
Covariates
The analysis included three models. In Model 1, associations were adjusted for sex, technical batch (cohort-specific variable) and estimated cell type proportions at birth, and additionally for child age in childhood and adolescence. The cell type proportions included CD8+ T-cells, CD4+ T-cells, natural killer cells, B cells, monocytes, granulocytes, and nucleated red blood cells at birth, estimated by using a cord blood-specific reference [10] and using the ‘Houseman method’ [11] using the Reinius reference set in peripheral blood [12]. Model 2 was additionally adjusted for maternal age, pre-pregnancy BMI, smoking (sustained smoking vs no smoking or stopping in early pregnancy), and gestational age at birth, to account for maternal prenatal factors. Model 3 additionally included offspring BMI and smoking (yes vs no). Models 1 and 2 were run at all three time points (birth, childhood, and adolescence) and model 3 in childhood and adolescence only, to account for offspring-specific covariates.
Statistical analysis
Cohort-specific epigenome-wide association analyses
The flow chart of the study design is given in Supplementary File Fig. 1, and analyses were described in a pre-specified analysis plan (Supplementary File). Cohorts used a common script to perform independent epigenome-wide linear regression analyses with robust standard errors in R.
Meta-analysis
To minimize human error, researchers from two centres independently performed quality control of the cohort-level results and fixed-effects inverse-variance weighted meta-analyses and verified the results. Single cohort CpGs and 44,960 cross-reactive CpGs were removed [13, 14]. The final results included 429 959 (birth), 429 233 (childhood), and 427 349 (adolescence) CpGs. Multiple testing burden was accounted for using the method of Benjamini and Hochberg [15] and setting FDR to 5%. We also assessed CpGs associations with a more stringent Bonferroni correction (P < 1 × 10−7). The nearest gene for all CpGs were annotated based on the Illumina annotation file. We assessed inter-study heterogeneity by the I2 statistic, and constructed forest plots to visualize the results for CpGs with I2 > 50%.
Sensitivity analyses
To investigate the robustness of our findings, several sensitivity analyses were performed for model 1 results. First, we ran a leave-one-study-out analysis for the CpGs with PFDR < 0.05 of each of the three age groups. Second, we re-ran the maternal meta-analyses for birth cohorts restricted to cohorts with participants of European ancestry only, which was the largest ancestry group (n = 9 501). Data in childhood and adolescence were only available for European ancestries. We examined overlap in the associated CpGs (PFDR < 0.05) of the three meta-analyses at birth, childhood, and adolescence to explore temporal persistence of differential methylation.
Enrichment analyses
We examined whether CpGs with I2 < 50% were enriched for CpGs previously identified at FDR-significance in the meta-analyses of EWASs of maternal folate concentrations [16], vitamin B12 concentrations, smoking [17], and pre-pregnancy BMI [18] using a hypergeometric test.
Functional analyses
To assess potential mechanisms linking MEA to offspring DNA methylation, we explored associations with gene expression, by comparing the associated CpGs at birth (at PFDR < 0.05) from model 1 with a catalogue containing 63 831 child-specific blood autosomal cis-expression quantitative trait methylation sites (cis-eQTMs, 1 Mb window) [19]. The GTEx gene-expression level of the identified nearest genes to the CpG sites were further assessed with the help of the webtool ‘Functional mapping and annotation of genetic associations’ (FUMA) [20]. We also explored whether the CpGs (PFDR < 0.05) were enriched in DNase I hypersensitive sites, commonly associated with regulatory regions, using eFORGE v2.0. with its default settings [21].
Results
Descriptive statistics
Descriptive statistics for the 37 datasets are shown in Table 1. The meta-analysis sample included 49.2% females. The mean number of years of MEA at the time of pregnancy ranged from 12.3 to 19 years. Cohort-specific distributions of MEA are shown in Supplementary Table 1. Mean maternal age ranged from 27.4 to 33.8 years. Maternal smoking during pregnancy prevalence ranged from 2% to 48%. Mean maternal pre-pregnancy BMI ranged from 22.3 to 28.0 kg/m2 and mean gestational age at birth from 38.5 to 40.2 weeks.
Meta-analyses of epigenome-wide association studies
Genomic inflation factors (λgc) for the models are shown in Supplementary Table 2. Figure 2 shows the Manhattan plots of model 1 at the three time points and Table 2 shows the top 20 significant hits (PFDR < 0.05) at birth and all hits for childhood and in adolescence. QQ plots of all the meta-analysis Manhattan plots for models 2 and 3 are reported in Supplementary File, Figs. 2 and 3. In model 1, MEA was associated with DNA methylation at 473 CpGs at birth, one CpG in childhood and four CpGs in adolescence at PFDR < 0.05 (Fig. 2, Table 2, and Supplementary Table 2 and 3). Using a more stringent Bonferroni-corrected p-value cut-off of P < 1 × 10−7, 182 CpGs at birth were associated, as well as all CpGs in childhood and in adolescence. cg25949550 (CNTNAP2) was the only CpG associated with MEA at all three time points. For each year increase in MEA, DNA methylation was higher by 0.05% (SE = 0.006, P ≤ 3.5 x 10−8, I2 = 41.5) at birth, 0.06% (SE = 0.006, P ≤ 7.6x10−8, I2 = 1.1) in childhood, and 0.08% (SE = 0.006, P ≤ 4.1 × 10−10, I2 = 37.7) in adolescence.

The x axis is the chromosomal position, and the y axis is the P-value on a -log10 scale. The blue line corresponds to the first CpG site for which PFDR < 0.05 and red line indicates suggestive significance P = 1 × 10−7. The Manhattan plot of the fully adjusted models are presented in Supplementary Fig. 2.
In the fully adjusted model 2 and 3 (Supplementary Table 4, 5 and Supplementary File, Fig. 2), MEA was associated with DNA methylation at two CpGs at birth, two in childhood and three in adolescence at PFDR < 0.05. These overlapped with CpGs found in model 1. Using a Bonferroni-corrected p-value cut-off of P < 1 × 10−7, DNA methylation at one CpG remained associated at birth, two in childhood and three in adolescence. Twenty-four CpGs had I2 > 50 at birth.
Sensitivity meta-analyses
The leave-one-out analyses on the 24 CpGs with I2 > 50 at birth showed for some of these (e.g., cg01952185, cg05383657), the meta-analysis results were influenced by the Generation R Study. However, removing this study resulted in larger absolute effect sizes, therefore any potential influence of Generation R would be towards the null (Supplementary File, Figures 4,5). Findings were consistent with our results at birth when only studies of European ethnicity were assessed (r = 0.97) (Supplementary Table 6).
Enrichment analysis
At birth, we observed enrichment (P < 1 × 10-5) for findings from previous EWASs of other prenatal exposures, namely maternal folate, vitamin B12 concentrations, smoking and pre-pregnancy BMI (Penrichment< range = 1.9 x 10−04 to 2.4 x 10−138) (Table 3). The directions of the effects were concordant for all the overlapping CpGs for maternal folate and vitamin B12 concentrations and were in the expected opposite direction for all the overlapping CpGs between MEA and maternal smoking (except for cg23989336, which was in the same direction) and pre-pregnancy BMI. For childhood and adolescence there was enrichment only for maternal smoking during pregnancy (Penrichment < 0.02 and 0.001).
Functional analyses
Using the CpGs suggestively associated with MEA at birth (at P < 1 × 10−5) from model 1, we found 89 unique CpG-gene expression pairs (cis-eQTMs) (P < 1 × 10−5) in an eQTM atlas based on blood samples collected in childhood (6–11 years). These cis-eQTMs involved 74 unique CpGs and 68 unique transcript clusters, which can be interpreted as putative genes (Supplementary Table 7). Increased DNA methylation was associated with decreased expression in 43 of these eQTMs, with increased expression in 46. Seventeen CpGs were associated with expression of HOTAIRM1 and six CpGs with expression of FRG1BP. We further assessed the tissue expression related to the genes of the identified 68 unique transcript clusters using GTEx gene-expression level in FUMA. The genes were found to be expressed across multiple tissues; however, multiple clusters of genes were observed in the brain and heart tissues (Supplementary File, Fig. 6). Using eFORGE (at P < 1 × 10−5) we found enrichment of DNAase I hypersensitive sites and of specific transcription factor motifs in adolescent blood (Supplementary File, Fig. 7).
Discussion
Our well-powered meta-analysis combining results from 37 studies from high income countries showed that MEA is associated with DNA methylation in the offspring at birth, in childhood, and in adolescence. Robust associations with MEA were found for 473 CpG sites at birth, one in childhood, and four in adolescence. At all ages, there was enrichment for findings from previous EWAS on maternal folate concentrations, vitamin B12 concentrations, smoking, and pre-pregnancy BMI.
Meta-analysis
DNA methylation at cg25949550 was consistently positively associated with MEA across all models and time points. A 1-year increase in MEA was associated with an increase of 0.05–0.08% in blood DNA methylation at cg25949550. This CpG is located at intron 1 of CNTNAP2, and overlaps with binding sites of transcription repressors SIN3A, CTBP2, CTCF and REST. CNTNAP2 genetic variations have been implicated in multiple neurodevelopmental disorders including schizophrenia, epilepsy, autism spectrum disorder, attention-deficit/hyperactivity disorder, and mental retardation [22]. Notably, in our study the associations of cg25949550 with MEA in pregnancy after adjusting for sustained maternal smoking during pregnancy disappeared in birth and childhood studies but remained in adolescents. DNA methylation at cg25949550 has been repeatedly found to be strongly associated with maternal smoking during pregnancy as well as with personal smoking in adults [23]. Among the MEA related CpG sites at birth, four CpGs, cg05575921 (AHRR), cg12803068 (MYO1G), cg22132788 (MYO1G) and cg21161138 (AHRR) overlapped with the findings from a previous large EWAS on own educational attainment by Linner et al. among adults. All these CpGs are strongly related to personal smoking and cg05575921 is one of the top CpGs related to smoking, was the strongest associated CpG site in both studies (P < 1 × 10−17). Linner et al. also found that all nine CpGs associated with the participant’s own educational attainment overlapped with those from the EWAS of maternal smoking, which is concordant with our study. Similarly, Van Dongen et al. identified that educational attainment CpGs overlapped with smoking signatures in a meta-analysis of four cohorts [24].
Consistent with previous studies assessing associations of socio-economic status with DNA methylation [4, 25], MEA associated CpGs at birth persisted only minimally in childhood and adolescence. Persistence of differential DNA methylation in offspring may not be a pre-requisite for long-term impacts of MEA on offspring health, as transient differential DNA methylation in utero can cause lasting functional changes predisposing offspring to later adverse outcomes [26,27,28]. We observed attenuation in the associations (models 2 and model 3) after adjusting for prenatal covariates such as maternal BMI, smoking, age, and gestational age. This was expected and emphasizes that the in-utero environment represents the combined effect of multiple prenatal factors. We are aware that there are other covariates that we were unable to adjust for in this study and which may affect the identified associations. However, we believe the covariates used were representative of several important aspects of the social dynamics of health.
Enrichment analysis
In the enrichment analysis, we observed that 85 of 473 CpGs overlapped with CpGs identified in relation to maternal smoking during pregnancy and had the expected opposite direction of effect for all the CpGs (except for cg23989336). Maternal smoking has repeatedly been found to be negatively associated with educational attainment: mothers with lower education are more likely to continue smoking in pregnancy compared to mothers with higher education [29]. A systematic review of 63 studies using Mendelian randomization identified robust evidence that higher educational attainment decreases smoking [30]. Gilman et al. evaluated a potential causal effect of educational attainment on smoking and observed that adjusting for a wide range of social factors had little impact on the association between the two [31]. It is therefore likely that smoking is rather a consequence, acting as mediator between educational attainment and health outcomes. Similarly, we observed overlap of sites associated with MEA with other prenatal exposures involved in in-utero programming such as maternal folate and vitamin B12 concentrations (with concordant directions of effect), and maternal pre-pregnancy BMI. The overlap of CpGs between MEA and these prenatal exposures may indicate a shared social molecular architecture between them leading to common biosocial pathways that may influence health outcomes, as often observed in observational studies.
Functional analyses
We found cis-eQTM involving the HOTAIRM1 and FRG1BP genes. Genetic variations in HOTAIRM1 are known to be involved in the neuronal differentiation and associated with waist-to-hip ratio phenotype. CpGs annotated to this gene were differentially methylated in newborns in relation to sustained maternal smoking during pregnancy and to own smoking in adults [23]. The HOTAIRM1gene also epigenetically controls the expression of the proneural transcription factor NEUROGENIN 2 that is critical for brain development [32]. FRG1BP (previously known as C20orf80, FRG1B) has been found to be associated with body weight, body height at birth and ocular sarcoidosis phenotypes [33]. Furthermore, we found enrichment of DNAase I hypersensitive sites and of specific transcription factor motifs in adolescents at RAR, ESRRA, V LXR and CTCF (Supplementary File, Fig. 7) regulating genes which play roles in cell differentiation, proliferation, have neuroprotective actions and regulate cholesterol metabolism, inflammation, autoimmunity, and cancer [34, 35]. Overall, these findings from our gene expression and tissue specific enrichment may indicate a role of MEA in important biological processes and pathways of the offspring, aligning with their multifaceted role observed in epidemiological studies.
Due to the multidimensionality of MEA, it has remained a challenge for researchers to disentangle the interrelationships with its close correlates including income, employment, and socio-economic status. These measures reflect different types of resources that may differentially impact a child’s biological development. Furthermore, our measure of educational attainment is unable to capture differences in educational quality, type, or other institutional or systemic factors that might independently influence biological health. It also focuses on individual-level aspects of education, leaving out the social context in which the education and health processes are embedded [36]. This raises several questions regarding the biological processes underlying these associations and our study should be seen as a steppingstone in this regard. Our findings likely represent a myriad of pathways related to MEA including adverse intrauterine (such as nutrition or toxicants), as well as childhood and adolescent exposures; thus, it is plausible that MEA is an upstream risk factor for proximal health behaviours. More research is warranted to understand the causality, to examine these associations in more ethnically diverse cohorts, and to study these associations in larger samples at later ages to gain in-depth insight into life-course trajectories.
Strengths and limitations
The main strength of this study is that it uses a large sample size and three critical time points of human development from birth up to adolescence. We harmonized MEA to promote comparability of results across all cohorts. The summary statistics from our study should be a useful resource for future studies to further examine the interplay of various social factors and their associations with numerous biological pathways. MEA captures various biosocial dimensions of health as highlighted by our enrichment analyses, and our examination of potentially related factors, such as maternal smoking, provides a platform for future studies to disentangle potential causal relationships.
Our findings should also be interpreted in the light of certain limitations. The participants in our study were relatively well-educated and from high income countries and thus, our findings may not be generalizable to disadvantaged populations that are more vulnerable to adverse health outcomes. This study included mostly individuals of European ancestry and a small sample from African and Hispanic backgrounds due to lack of data availability; hence, the findings are not generalizable to ancestries beyond Europeans. We assessed MEA at the time of pregnancy and did not investigate education attained later in the childhood and adolescent cohorts. It is important to emphasize that we did not aim to draw direct causal conclusions, or to distinguish how much of these associations were confounded by other factors such as paternal education to understand the importance of maternal factors in the context of the family on DNA methylation [37]. Importantly, we observed overlap of methylation sites between maternal smoking and education, and the adjustment for sustained maternal smoking attenuated the associations at birth. It is likely that among individuals who continue to smoke throughout pregnancy, those of lower educational status might be over-represented.
We found that MEA at the time of pregnancy was associated with offspring DNA methylation at birth, in childhood, and in adolescence. The findings from the gene expression and enrichment analyses identified differential DNA methylation of genes involved in important biological processes. This may mean that socio-economic factors such as maternal education leave a “biological residue” which in turn may influence development, health, and wellbeing. Given the known association between higher maternal educational attainment and unhealthy maternal conditions (for ex. increased BMI, history of smoking, low folate levels, low Vitamin B12) [38] that have been linked to differences in DNA methylation patterns, investing in education access, especially in low-resource settings, holds potential to reduce health inequalities and improve the well-being across generations. This is consistent with the hypothesis that public health benefits are gained by improving educational attainment and addressing the social determinants of health [36, 39]. The summary statistics from this study provide an important resource for future studies to further investigate the intricate biosocial pathways involved in in-utero programming and establish a more comprehensive understanding of intergenerational health.
Disclaimer
Where authors are identified as personnel of the International Agency for Research on Cancer/ World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/ World Health Organization.
Data availability
Meta-analysis results files will be deposited in the EWAS Catalogue data repository (http://ewascatalog.org) upon publication.
References
Jackson MI, Kiernan K, McLanahan S. Maternal education, changing family circumstances, and children’s skill development in the United States and UK. Ann Am Acad Pol Soc Sci. 2017;674:59–84.
Boyle MH, Racine Y, Georgiades K, Snelling D, Hong S, Omariba W, et al. The influence of economic development level, household wealth and maternal education on child health in the developing world. Soc Sci Med. 2006;63:2242–54.
Cerutti J, Lussier AA, Zhu Y, Liu J, Dunn EC. Associations between indicators of socioeconomic position and DNA methylation: a scoping review. Clin Epigenet. 2021;13:221.
Alfano R, Guida F, Galobardes B, Chadeau-Hyam M, Delpierre C, Ghantous A, et al. Socioeconomic position during pregnancy and DNA methylation signatures at three stages across early life: epigenome-wide association studies in the ALSPAC birth cohort. Int J Epidemiol. 2019;48:30–44.
Karlsson Linnér R, Marioni RE, Rietveld CA, Simpkin AJ, Davies NM, Watanabe K, et al. An epigenome-wide association study meta-analysis of educational attainment. Mol Psychiatry. 2017;22:1680–90.
Felix JF, Joubert BR, Baccarelli AA, Sharp GC, Almqvist C, Annesi-Maesano I, et al. Cohort Profile: Pregnancy And Childhood Epigenetics (PACE) Consortium. Int J Epidemiol. 2018;47:22–23u.
UNESCO. ISCED 1997. International Standard Classification of Education. http://uis.unesco.org/sites/default/files/documents/international-standard-classification-of-education-1997-en_0.pdf.
Lehne B, Drong AW, Loh M, Zhang W, Scott WR, Tan S-T, et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015;16:37.
Tukey JW. Exploratory data analysis. Addison-Wesley; (1977).
Gervin K, Salas LA, Bakulski KM, van Zelm MC, Koestler DC, Wiencke JK, et al. Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin Epigenet. 2019;11:125.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 2012;13:86.
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361.
Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9.
Naeem H, Wong NC, Chatterton Z, Hong MKH, Pedersen JS, Corcoran NM, et al. Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genom. 2014;15:51.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Joubert BR, den Dekker HT, Felix JF, Bohlin J, Ligthart S, Beckett E, et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun. 2016;7:10577.
Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, et al. DNA methylation in newborns and maternal smoking in pregnancy: Genome-wide Consortium Meta-analysis. Am J Hum Genet. 2016;98:680–96.
Sharp GC, Salas LA, Monnereau C, Allard C, Yousefi P, Everson TM, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet. 2017;26:4067–85.
Exposome-omics-Wide Association Study (ExWAS). (2020). https://helixomics.isglobal.org/#.
Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1–10.
Breeze CE, Reynolds AP, van Dongen J, Dunham I, Lazar J, Neph S, et al. eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data. Bioinformatics. 2019;35:4767–9.
Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet. 2014;22:171–8.
Sikdar S, Joehanes R, Joubert BR, Xu C-J, Vives-Usano M, Rezwan FI, et al. Comparison of smoking-related DNA methylation between newborns from prenatal exposure and adults from personal smoking. Epigenomics. 2019;11:1487–1500.
van Dongen J, Bonder MJ, Dekkers KF, Nivard MG, van Iterson M, Willemsen G, et al. DNA methylation signatures of educational attainment. NPJ Sci Learn. 2018;3:7.
Laubach ZM, Perng W, Cardenas A, Rifas-Shiman SL, Oken E, DeMeo D, et al. Socioeconomic status and DNA methylation from birth through mid-childhood: a prospective study in Project Viva. Epigenomics. 2019;11:1413–27.
Cosin-Tomas M, Cilleros-Portet A, Aguilar-Lacasaña S, Fernandez-Jimenez N, Bustamante M. Prenatal maternal Smoke, DNA methylation, and multi-omics of tissues and child health. Curr Environ Heal Rep. 2022;9:502–12.
Rauschert S, Melton PE, Burdge G, Craig JM, Godfrey KM, Holbrook JD, et al. Maternal smoking during pregnancy induces persistent epigenetic changes into adolescence, independent of postnatal smoke exposure and is associated with cardiometabolic risk. Front Genet. 2019;10.
Wiklund P, Karhunen V, Richmond RC, Parmar P, Rodriguez A, De Silva M, et al. DNA methylation links prenatal smoking exposure to later life health outcomes in offspring. Clin Epigenet. 2019;11:97.
Kandel DB, Griesler PC, Schaffran C. Educational attainment and smoking among women: Risk factors and consequences for offspring. Drug Alcohol Depend. 2009;104:S24–33.
Treur JL, Munafò MR, Logtenberg E, Wiers RW, Verweij KJH. Using Mendelian randomization analysis to better understand the relationship between mental health and substance use: a systematic review. Psychol Med. 2021;51:1593–624.
Gilman SE, Martin LT, Abrams DB, Kawachi I, Kubzansky L, Loucks EB, et al. Educational attainment and cigarette smoking: a causal association? Int J Epidemiol. 2008;37:615–24.
Rea J, Menci V, Tollis P, Santini T, Armaos A, Garone MG, et al. HOTAIRM1 regulates neuronal differentiation by modulating NEUROGENIN 2 and the downstream neurogenic cascade. Cell Death Dis. 2020;11:527.
GeneCards: The Human gene database. https://www.genecards.org/. Accessed 28 April (2022).
The Nuclear Receptor FactsBook. Elsevier; (2002).
Bilotta MT, Petillo S, Santoni A, Cippitelli M Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front Immunol. 2020;11.
Zajacova A, Lawrence EM. The relationship between education and health: reducing disparities through a contextual approach. Annu Rev Public Health. 2018;39:273–89.
Sharp GC, Lawlor DA, Richardson SS. It’s the mother!: How assumptions about the causal primacy of maternal effects influence research on the developmental origins of health and disease. Soc Sci Med. 2018;213:20–27.
Viinikainen J, Bryson A, Böckerman P, Kari JT, Lehtimäki T, Raitakari O, et al. Does better education mitigate risky health behavior? A mendelian randomization study. Econ Hum Biol. 2022;46:101134.
WHO. Social determinants of health. https://www.who.int/health-topics/social-determinants-of-health#tab=tab_1.
Acknowledgements
This work received support from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 733206 (LifeCycle), grant agreement no. 873749 (LongITools), and grant agreement no. 848158 (EarlyCause). The funding organisations were not involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. The Supplementary File contains detail of the cohort specific acknowledgements and fundings.
Funding
Open Access funding provided by University of Oulu (including Oulu University Hospital).
Author information
Authors and Affiliations
Contributions
SS, PC, and JF conceptualised and designed the study. PC, SS, and JF prepared the original protocol. PC, SS, JF, GSM, VK, and MK coordinated the project. PC, GSM, and VK did the data management and analysis. PC and GSM led the quality control of data. PC, SS, JF, VK, GSM, and MK wrote the manuscript with input from all co-authors. All authors contributed to the interpretation of the findings. All authors critically revised the paper for intellectual content and approved the final version of the manuscript. All authors had full access to all the data in the study, although not all authors will have accessed the totality of the data; rather, they will have accessed their own site data. PC, GSM, SS, JF and VK take responsibility for the integrity of the data and the accuracy of the data analysis. All authors had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choudhary, P., Monasso, G.S., Karhunen, V. et al. Maternal educational attainment in pregnancy and epigenome-wide DNA methylation changes in the offspring from birth until adolescence. Mol Psychiatry (2023). https://doi.org/10.1038/s41380-023-02331-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41380-023-02331-5
