
Abstract
Chronic myelomonocytic leukemia is characterized by persistent absolute monocytosis (≥1 × 109/l) in the peripheral blood and dysplasia in ≥1 lineages. In the absence of dysplasia, an acquired clonal genetic abnormality is required or causes for reactive monocytosis have to be excluded. Oligomonocytic chronic myelomonocytic leukemia showing increased monocytes but no absolute monocytosis in the peripheral blood occurs occasionally. These cases are likely classified as myelodysplastic syndrome or myelodysplastic/myeloproliferative neoplasm, unclassifiable. A subset eventually develop overt chronic myelomonocytic leukemia. Better characterization of oligomonocytic chronic myelomonocytic leukemia is essential since the distinction between chronic myelomonocytic leukemia and myelodysplastic syndrome is clinically relevant. We identified 44 cases of oligomonocytic chronic myelomonocytic leukemia (≥10% peripheral blood monocytes with absolute monocyte count of 0.5–1 × 109/l) and 28 consecutive chronic myelomonocytic leukemia controls. Clinicopathologic features were compared and mutation analysis was performed. Oligomonocytic chronic myelomonocytic leukemia patients were significantly younger (median age of 65 vs 72). They had lower WBC and absolute neutrophil count, while the monocyte percentage, hemoglobin and platelet counts were similar in the two groups. The myeloid to erythroid ratio was predominantly decreased or normal, compared with the characteristic increase in chronic myelomonocytic leukemia (P=0.006). 38% of patients progressed to overt chronic myelomonocytic leukemia (median: 12 months). The overall percentage of mutations was significantly lower in oligomonocytic chronic myelomonocytic leukemia. However, the most frequent mutations in both groups were the ‘signature’ chronic myelomonocytic leukemia mutations in ASXL1, TET2 and SRSF2. Mutations in CBL were found exclusively in overt chronic myelomonocytic leukemia. In conclusion, we demonstrate clinical and genetic similarities between overt chronic myelomonocytic leukemia and oligomonocytic chronic myelomonocytic leukemia. The findings suggest that at least a subset of oligomonocytic chronic myelomonocytic leukemia represents early phase ‘dysplastic type’ chronic myelomonocytic leukemia.
Similar content being viewed by others

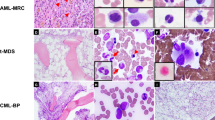
Main
Based on the French–American–British (FAB) classification scheme for myeloid disorders published in 1976, chronic myelomonocytic leukemia was considered to belong to the myelodysplastic syndrome category, because of presence of dysplasia, frequently elevated bone marrow blast count similar in range to those seen in cases of myelodysplastic syndrome with excess blasts and a similar prognosis.1 The FAB classification defined chronic myelomonocytic leukemia as the presence of an absolute monocytosis (over 1 × 109/l), ‘often associated with an increase in mature granulocytes with or without evidence of dysgranulopoiesis’.1 The cut-off value of 1 × 109/l absolute monocytosis in peripheral blood was determined based on experience and expert consensus, and has since been universally accepted.2
At that time it was unclear how to account for chronic myelomonocytic leukemia cases with high white blood cell (WBC) counts. Therefore in 2001 the World Health Organization (WHO) classification included chronic myelomonocytic leukemia into a newly created myelodysplastic/myeloproliferative neoplasm category.3 The WHO did not make significant changes to the other diagnostic criteria of chronic myelomonocytic leukemia.3 Based on the most recent WHO Classification update, chronic myelomonocytic leukemia is defined by persistent peripheral blood monocytosis ≥1 × 109/l, with monocytes accounting for ≥10% of the WBC, accompanied by dysplasia in ≥1 lineages. In the absence of dysplasia, an acquired clonal cytogenetic/molecular abnormality is required or causes for reactive monocytosis have to be excluded.4, 5
In recent years, much progress has been made in understanding the molecular pathogenesis of myeloid neoplasms, including chronic myelomonocytic leukemia. Clonal cytogenetic abnormalities are found in 20–40% of patients, but none is specific. On the other hand, certain gene mutations are enriched in chronic myelomonocytic leukemia cases. As many as 40% of patients exhibit point mutations of NRAS/KRAS genes at diagnosis, or during the course of disease and the presence of mutations in both TET2 and SRSF2 has been found to be strongly associated with the chronic myelomonocytic leukemia diagnosis.6, 7, 8, 9 In fact, either TET2, SRSF2 and/or ASXL1 gene mutation are present in 90% of patients.8 Mutations in SRSF2 and ASXL1, each occur at ~50% of cases, which is much higher than the frequency with which either mutation is seen in myelodysplastic syndrome (<20% of cases).10, 11 Mutations in signaling pathway genes like JAK2, NRAS and CBL are more common in chronic myelomonocytic leukemia and rare in myelodysplastic syndrome.6, 12 Thus, in problematic cases in which the monocyte count is borderline, raising the differential diagnosis of myelodysplastic syndrome, a characteristic molecular profile may help to support the diagnosis of chronic myelomonocytic leukemia. In addition, the presence of NRAS, RUNX1, SETBP1 or ASXL1 mutations in chronic myelomonocytic leukemia is associated with a more aggressive course independent of the cytogenetic abnormalities.8, 12, 13, 14, 15
We have encountered occasional cases of myeloid neoplasms showing increased peripheral blood monocytes (≥0.5 × 109/l) and relative monocytosis (≥10%), but not at the level of absolute monocytosis required by the WHO classification. A subset of these cases also had increased bone marrow monocytes. Clinically and pathologically, these cases did not fit any well-defined WHO category and were usually classified as myelodysplastic syndrome or as myelodysplastic/myeloproliferative neoplasm, unclassifiable. Better characterization of these cases is essential, as the distinction between chronic myelomonocytic leukemia and myelodysplastic syndrome is clinically relevant: chronic myelomonocytic leukemia is currently considered as a clinically and genetically distinct entity with a unique clinical presentation, natural history and a generally poor prognosis. The aim of the study was to determine if these ‘oligomonocytic’ cases were clinically and genetically compatible with an early stage of chronic myelomonocytic leukemia or represented a different subtype of myeloid neoplasm.
Materials and methods
Patients
We searched the pathology archives at six academic institutions in the United States: Weill Cornell Medical College, MD Anderson Cancer Center, Stanford University Medical Center, Cleveland Clinic, Massachusetts General Hospital and the Hospital of the University of Pennsylvania for bone marrow biopsies diagnosed as de novo myeloid neoplasms with ≥10% monocytes in PB with absolute monocyte count of ≥0.5 but <1 × 109/l (based on Dusseldorf registry data)16 and/or ≥10% monocytes in BM. Alpha naphtyl acetate esterase or alpha naphtyl butyrate esterase (alone or in combination with naphthol-ASD-chloroacetate esterase) and/or immunohistochemistry for CD14 were used in selected cases to aid in quantifying monocytes on the marrow aspirate and biopsy respectively. All included cases also had to meet the remaining 2008 WHO Classification requirements for diagnosis of chronic myelomonocytic leukemia. Based on the 2016 revision to the WHO, the cases were further classified as ‘proliferative type’ (WBC count ≥13 × 109/l) and ‘dysplastic type’ (WBC <13 × 109/l).4 In addition, 28 consecutive cases of overt chronic myelomonocytic leukemia, diagnosed in accordance with the WHO criteria were identified in the pathology archives of Weill Cornell Medical College to serve as controls. Chronic myelomonocytic leukemia cases were classified as CMML-0 (<2% blasts in peripheral blood and <5% blasts in bone marrow); CMML-1 (2–4% blasts in peripheral blood and/or 5–9% blasts in bone marrow); and CMML-2 (5–19% blasts in peripheral blood, 10–19% in bone marrow, presence of Auer rods). Cases of therapy-related myeloid neoplasm defined as history of prior cytotoxic chemotherapy or radiation with significant exposure to hematopoietic marrow were excluded from the study. Chronic myelomonocytic leukemia-specific prognostic scoring system (CPSS) was applied to predict survival.15 Briefly, it incorporates a genetic score (evidence of ASXL1, NRAS, RUNX1, SETBP1 mutations and cytogenetic risk group) with presence of increased bone marrow blasts, increased WBC count and RBC transfusion dependency. Clinical information was retrieved from the electronic medical records. This study was approved by the Institutional Review Boards of all participating institutions.
Cytogenetics and Fluorescence In Situ Hybridization
Conventional cytogenetic analysis was performed on G-banded metaphase cells prepared from unstimulated bone marrow aspirate cultures using standard techniques. Twenty metaphases were analyzed and the results were reported using the International System for Human Cytogenetic Nomenclature. Fluorescence in situ hybridization and/or molecular genetic methods for detecting BCR-ABL1, and PDGFRA or PDGFRB translocations were performed at respective institutions as part of the routine clinical work-up.
Bone Marrow Morphologic and Histologic Assessment
The evaluated histological criteria were agreed by all participants prior to case collection to ensure consistency in morphological assessment. The bone marrow aspirate smears and core biopsy either obtained at the time of diagnosis or the first bone marrow procedure ever performed with paired peripheral blood smears were reviewed by at least one observer at each institution. A differential count based on at least 200 cells (or all available cells if <200 present) was performed, and ring sideroblasts were assessed on Prussian blue or Perls-stained bone marrow aspirate smears. All histological review was blinded to next-generation sequencing testing results.
Targeted Next-Generation Sequencing
Targeted next-generation sequencing was performed on DNA samples extracted from frozen unfractionated bone marrow cells collected at the time of diagnosis. In a subset of cases, DNA was extracted from unstained and unfixed bone marrow aspirate smears. Briefly, genomic DNA was fragmented to 3–4 kb by shearing using QSonica (Newtown, CT) Q800R2 instrument according to the manufacturer’s recommendations. Targeted enrichment of 45 genes (ABL1, ASXL1, BCOR, BRAF, CALR, CBL, CEBPA, DNMT3A, ETV6, EZH2, FAM5C, FLT3, GATA1, GATA2, HNRNPK, IDH1, IDH2, IKZF1, JAK1, JAK2, KDM6A, KIT, KRAS, MPL, NFE2, NOTCH1, NPM1, NRAS, PHF6, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SH2B3, SMC1A, SMC3, SRSF2, STAG2, SUZ12, TET2, TP53, U2AF1, WT1, ZRSR2) recurrently mutated in myeloid malignancies was performed using the Thunderstorm system (Raindance Technologies, Billerica, MA) using a custom primer panel followed by sequencing using the Illumina MiSeq (v3 chemistry) yielding 260-bp paired end reads. Sequence adapters and low quality reads were excluded or trimmed using Trimmomatic followed by mapping to the human reference genome (hs37d5) using BWA MEM removing both discordant and non-uniquely mapping alignments. Variant calling was performed using VarDict amplicon mode excluding variants in low complexity and PCR primer regions.17 Variants were subsequently annotated using SnpEff 4.1.18 Common SNPs likely to be non-somatic with minor allele frequency greater than 0.25% were excluded from subsequent analysis with the exception of variants identified at least twice in the Catalog of Somatic Mutations in Cancer (COSMIC) v68.19
Statistical Analyses
For continuous variables, data are reported as median and range. For nominal variables, data are reported as the number of patients unless otherwise specified. Disease-specific survival was calculated from the day of diagnosis to the last follow-up or death attributed to chronic myelomonocytic leukemia or disease progression. Patients who received hematopoietic stem cell transplant were censored at the time of the procedure. Kaplan–Meier estimator was used to estimate survival probability. Survival difference between groups was tested by log-rank test. Fisher’s exact and Chi-square tests were used for categorical comparisons. No adjustments for multiplicity were made. Wilcoxon Rank-sum test was used for the comparison of number of mutations detected in each of the two groups. The frequency of mutations in each gene was compared between the two groups by Fisher’s exact test. To determine the likelihood that mutations in the two genes are mutually exclusive or co-occurring across the cases, the relationship between mutations of the two genes was tested by a Fisher’s exact test and the Odds Ratio (OR) was calculated. This guideline was used to classify the nature of the relationship: 0<OR<0.1: strongly mutual exclusive; 0.1<OR<0.5: some tendency toward mutual exclusive; 0.5<OR<2: no association; 2<OR<10: tendency towards co-occurrence; 10<OR: strong tendency towards co-occurrence. The statistical test is two-sided and an alpha level of 0.05 was used to determine the statistical significance. All analyses were performed in SAS9.4 (SAS Institute, Cary, NC, USA).
Results
Clinical Features
Forty four patients satisfied the criteria for oligomonocytic chronic myelomonocytic leukemia (Table 1). There were 33 men and 11 women with a median age of 65 (range 31–87) years. The group of 28 control chronic myelomonocytic leukemia patients had an older median age of 72 (range 58–88) years (P=0.004). At presentation, LDH was increased in 71% of oligomonocytic chronic myelomonocytic leukemia patients with the average of 376 U/l (range, 104–1197). 14% of patients had organomegaly. Mean WBC at presentation was 3.9 × 109/l (range, 1.8–9.4) in the oligomonocytic chronic myelomonocytic leukemia patients, compared with 17.2 × 109/l (range, 3.0 to 69; P<0.001) in all control patients. However, when the ‘dysplastic’ chronic myelomonocytic leukemia group was separated from the controls, the WBC values were not statistically different (3.9 vs 7.4 × 109/l). The mean peripheral blood monocyte percentage at presentation was 16.8% (range, 10–48%) in the oligomonocytic group, and was similar compared with 25.2% in the control group (range, 10–47%, P=0.07). Predictably, the absolute monocyte count was significantly lower in the oligomonocytic group, compared with controls (0.75 vs 4.15 × 109/l, P<0.001), but was similar to the ‘dysplastic’ control group (0.75 vs 1.9). 23/43 (53%) of oligomonocytic chronic myelomonocytic leukemia patients presented with neutropenia, compared with 1/28 control patients (P<0.001). Anemia was the most common cytopenia at presentation in the oligomonocytic group (73% patients) with the mean hemoglobin of 10.0 g/dl, very similar to the control group with the mean hemoglobin of 10.9 g/dl. Elevated MCV was present in 37% of the oligomonocytic chronic myelomonocytic leukemia patients with the mean MCV of 97 fL (range, 80–120.9), similar to control cases. Compared with overt chronic myelomonocytic leukemia, patients with oligomonocytic chronic myelomonocytic leukemia were slightly less likely to present with thrombocytopenia (63 vs 89%). The mean platelet values were similar at 138 vs 103 × 109/l. These results were similar, when the oligomonocytic cases were compared with ‘dysplastic type’ controls alone.
16/42 (38%) of oligomonocytic patients developed overt chronic myelomonocytic leukemia after a median follow-up of 12 (range, 1–84) months. 11/42 (26%) of oligomonocytic patients developed acute myeloid leukemia after a median follow-up of 10 (range, 1–72) months after the initial presentation. 3/11 patients (27%) had an intermediate stage of overt chronic myelomonocytic leukemia, while 8 patients progressed directly to acute leukemia. The incidence of progression to acute leukemia was similar to the control group (5/28 (18%), P=0.6). In the oligomonocytic group, there was no statistically significant association between development of chronic myelomonocytic leukemia or acute myeloid leukemia and history of prior cytotoxic chemotherapy or radiation treatment (data not shown).
A significant proportion of patients were closely followed and were not treated (21/42 oligomonocytic (50%) and 10/28 overt chronic myelomonocytic leukemia (36%) patients). Hypomethylating agents were the most common treatment choice for oligomonocytic chronic myelomonocytic leukemia (15/42, 36%), while combination chemotherapy was the most frequent choice for overt chronic myelomonocytic leukemia patients (8/28, 29%). 7/44 (16%) oligomonocytic patients and 4/28 (14%) controls received allogeneic stem cell transplant. There was no statistical difference between therapy regimens used. 19/44 oligomonocytic patients (43%) died of disease and 25 (57%) were alive (including 4 patients in remission following stem cell transplant) after a median follow-up of 19 (range, 1–132) months. In comparison, 5 (18%) patients in the control group died of disease, while 23 (82%) were alive (including 3 patients in remission post stem cell transplant) after a similar mean follow-up of 30 (range, 7–108) months. Median survival time with 95% confidence interval was 72.0 (17.0–108.0) months for oligomonocytic, while it was not reached for overt chronic myelomonocytic leukemia patients. The 5 year survival rate was 57.8% for oligomonocytic chronic myelomonocytic leukemia and 80.1% for control patients (log-rank test, P=0.027).
Morphologic and Cytogenetic Findings
Bone marrow cellularity was increased for age in 76% of the oligomonocytic chronic myelomonocytic leukemia patients. The myeloid to erythroid ratio was decreased in 50% of patients and increased in 32% of patients (Table 2). In contrast, most of the control patients had a high myeloid to erythroid ratio (68%, with a mean of 4.6; P=0.006). Thus, the median myeloid to erythroid ratio was lower in the oligomonocytic group compared with controls (1.9 vs 4.2), even though the mean and the range of myeloid to erythroid ratio was similar in the two groups. 26% of oligomonocytic cases had MF-2/MF-3 marrow fibrosis, compared with 18% of controls. The mean bone marrow monocyte count in the oligomocytic cohort was 9% (range, 0–25%), similar to 12% (range, 1–30%) in the control group. Bone marrow monocytes were increased in 70% of oligomonocytic chronic myelomonocytic leukemia patients; 41% had >10% monocytes. Myeloid dysplasia was present in 24 (55%) of oligomonocytic patients, erythroid dysplasia in 26 (59%) and megakaryocytic dysplasia in 33 (75%). Rare ring sideroblasts were seen in 15/37 (41%) of patients; 7 (19%) patients had >15% ring sideroblasts. These results were similar to BM findings in the control patients, including ‘dysplastic type’ control group (data not shown).
Oligomonocytic chronic myelomonocytic leukemia cases were most commonly diagnosed as refractory cytopenia with multilineage dysplasia (17 cases, 39%), followed by refractory anemia with excess blasts (13 cases, 30%), low-grade myelodysplastic syndrome (4 cases, 9%), myelodysplastic/myeloproliferative neoplasm unclassifiable (3 cases, 7%), refractory anemia with ring sideroblasts (3 cases, 7%), and one case each of ‘myeloid neoplasm’; unclassifiable myelodysplastic syndrome; chronic myelomonocytic leukemia and myelodysplastic syndrome with isolated del(5q). The control cases included CMML-0 (19 cases, 68%); CMML-1 (7 cases, 25%) and CMML-2 (2 cases, 7%). Following the analogy with chronic myelomonocytic leukemia, the oligomonocytic cases had a similar distribution of cases when separated into oligomonocytic CMML-0 (29/43 cases, 67%), oligomonocytic CMML-1 (5/43, 9%) and oligomonocytic CMML-2 (8/43, 19%).
Cytogenetic analysis demonstrated an abnormal karyotype in 34% of oligomonocytic patients. The most common abnormalities included monosomy 7/del(7q) (6 cases). Complex karyotype was seen in 3 cases. These findings were not significantly different from control patients (Table 2).
Next-Generation Sequencing Status
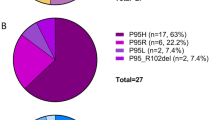
Adequate material was available for sequencing in 24/44 oligomonocytic and 25/28 control chronic myelomonocytic leukemia cases, including 11/16 ‘dysplastic type’ chronic myelomonocytic leukemia. In line with the published literature,4 the three most frequently mutated genes in the control group were TET2 (76% overall/70% in ‘dysplastic type’), ASXL1 (44%/45% ‘dysplastic type’) and SRSF2 (44%/20% ‘dysplastic type’). 40% (20% ‘dysplastic type’) patients had concurrent TET2 and SRSF2 mutations (Table 2 and Figure 1). Other genes that were mutated at lower frequency but above 10% included CBL (28%), RUNX1 (24%) and NRAS (24%). There were no significant molecular differences between ‘dysplastic type’ and ‘proliferative type’ chronic myelomonocytic leukemia. For the oligomonocytic group, the three most frequently mutated genes were also TET2 (46%), ASXL1 (21%) and SRSF2 (17%). 12.5% patients had concurrent TET2 and SRSF2 mutations. Other genes that were mutated at lower frequency but above 10% included NRAS (13%), TP53 (13%), U2AF1 (13%) and DNMT3A (13%) (Table 2 and Figure 2). SRSF2 alterations in both groups were either missense mutation or small deletion at the hotspot P95.

Mutation patterns observed in control chronic myelomonocytic leukemia cases. The plot represents the distributions of somatic lesions in genes mutated in at least one sample. Each column represents an individual patient sample. Red: mutation detected; green: mutation not detected; gray: mutation not tested.

Mutation patterns observed in oligomonocytic chronic myelomonocytic leukemia cases. The plot represents the distributions of somatic lesions in genes mutated in at least one sample. Each column represents an individual patient sample. Red: mutation detected; green: mutation not detected; gray: mutation not tested.
Overall, patients with chronic myelomonocytic leukemia had a higher percentage of mutated genes (relative to the number of genes tested) compared with oligomonocytic chronic myelomonocytic leukemia (11 vs 7%, P=0.023). Although the same genes: TET2, ASXL1 and SRSF2 represent the three most frequently mutated genes in both groups, chronic myelomonocytic leukemia patients had a significantly higher frequency of TET2 mutation (P=0.042). There was also an increased presence of CBL mutation in control patients with none detected in the studied oligomonocytic cases (P=0.002). Three of 7 CBL mutations co-occurred with SRSF and TET2; 2 of these 3 also had ASXL1 mutation. The other 4 CBL mutated cases had ASXL1 mutations +/− TET2 mutations.
Risk Assessment According to Chronic Myelomonocytic Leukemia-Specific Prognostic Scoring System (CPSS)
The comparison was performed according to the recently published CPSS-mol.15
Patients in oligomonocytic and control groups had a very similar case distribution with 45 vs 46% of patients in the low/intermediate-1 risk groups and 55 vs 54% in the intermediate-2/high risk groups, respectively. Patient survival in the oligomonocytic group was significantly different based on CPSS-mol with a median survival of 10.0 months in the high risk group, compared with 72.0 months in the intermediate-2 group, while it was not reached for low and intermediate-1 groups (P<0.001). The survival difference between oligomonocytic patients grouped in low/intermediate-1 risk and intermediate-2/high risk groups was also statistically significant (17 months vs not reached, P=0.027, see Figure 3a), as was survival for control CMML patients (25 months vs not reached, P=0.021, see Figure 3b).

(a) Kaplan–Meier curve of overall survival in the oligomonocytic chronic myelomonocytic leukemia cohort classified into CPSS-Mol risk groups. The number of patients in each category is as follows: 12 (55%) high and intermediate-2 risk group; 10 (45%) low and intermediate-1 risk group. Survival difference among groups was tested by log-rank test (P=0.027). (b) Kaplan–Meier curve of overall survival in the control chronic myelomonocytic leukemia cohort classified into CPSS-Mol risk groups. The number of patients in each category is as follows: 13 (54%) high and intermediate-2 risk group; 9 (46%) low and intermediate-1 risk group. Survival difference among groups was tested by log-rank test (P=0.021).
Discussion
For historical reasons, the diagnosis of chronic myelomonocytic leukemia required presence of an absolute monocytosis over 1 × 109/l. However, occasional cases of myeloid neoplasms fit all the chronic myelomonocytic leukemia criteria and show increased peripheral blood monocytes, but not in the range defined by the current classification schemes. These cases do not fit any well-defined WHO category and are likely to be classified as myelodysplastic syndrome, myelodysplastic/myeloproliferative neoplasm unclassifiable or receive a descriptive diagnosis. We have provisionally called them oligomonocytic chronic myelomonocytic leukemia. The aim of the study was to determine if oligomonocytic chronic myelomonocytic leukemia cases were clinically and genetically compatible with an early stage of chronic myelomonocytic leukemia or represented a different subtype of myeloid neoplasm. We also hope to provide guidance in refining the diagnostic guidelines for chronic myelomonocytic leukemia.
Anemia was the most common clinical presentation in oligomonocytic chronic myelomonocytic leukemia. In contrast to the patients with overt chronic myelomonocytic leukemia, all patients with oligomonocytic chronic myelomonocytic leukemia were of ‘dysplastic type’ with normal or decreased WBC and thus significantly lower absolute monocyte count, although the percentage of peripheral blood and bone marrow monocytes was similar between the two groups. In the control group, 2/3 of the cases were ‘dysplastic’ and 1/3 were ‘proliferative’ type.4 It appears that the presence of ‘proliferative’ type in the control group allowed for certain skewing of these results, since when the laboratory parameters were compared between oligomonocytic and ‘dysplastic’ type chronic myelomonocytic leukemia only, there were no significant differences. Bone marrow examination of oligomonocytic group showed that most patients had a low/normal myeloid to erythroid ratio, compared with control patients with significantly increased myeloid to erythroid ratio. Oligomonocytic chronic myelomonocytic leukemia patients appeared to have an aggressive clinical course, with 38% rate of disease progression to chronic myelomonocytic leukemia, 26% progression to acute myeloid leukemia and significant patient mortality. In this limited cohort, overall survival of oligomonocytic chronic myelomonocytic leukemia patients appeared significantly shorter compared with control patients. Based on the literature, the expected median overall survival in chronic myelomonocytic leukemia is 20–40 months, which is much shorter than the observed median overall survival for oligomonocytic chronic myelomonocytic leukemia patients in this study (72 months).15, 20, 21, 22 The reason for the observed significantly higher survival in the control chronic myelomonocytic leukemia patients compared with the published data and to the oligomonocytic cohort is unclear. Even though patients appeared well matched in all other regards, it is possible that the chosen controls may not have been entirely representative. It is also possible that oligomonocytic chronic myelomonocytic leukemia patients, at least at our institutions, may have benefitted from chronic myelomonocytic leukemia -directed therapy.
Although TET2, ASXL1 and SRSF2 mutations can also be found in other myeloid neoplasms, the observation that the same three genes are mutated at the highest frequency in both groups suggests biologic similarities. Concurrent mutations in TET2 and SFSR2 have been considered to be relatively specific for chronic myelomonocytic leukemia.23 Both groups showed co-occurrence of TET2 and SRSF2 mutations. This finding provides additional molecular evidence that at least a subset of oligomonocytic chronic myelomonocytic leukemia have pathogenic similarities to chronic myelomonocytic leukemia. Furthermore, all observed SRSF2 mutations involved the hotspot P95, further suggesting the pathogenic similarities between the two groups.
There were also molecular differences that may account for the clinicopathologic differences. Although the percentage of patients with TET2 and/or ASXL1 is similar (80 vs 67%), TET2 mutation was significantly less frequent in oligomonocytic chronic myelomonocytic leukemia. This finding suggests that TET2-mediated DNA methylation may be more effective in driving the chronic myelomonocytic leukemia phenotype compared with the other epigenetic regulator ASXL1. SRSF2 is considered relatively more specific for chronic myelomonocytic leukemia, occurring in 40 to 50% of cases23 but only in 10–15% cases of myelodysplastic syndrome24, 25 or other myeloid neoplasms. The frequency of SRSF2 mutations in the oligomonocytic cohort appeared to be in between what is described in the literature for conventional myelodysplastic syndrome and chronic myelomonocytic leukemia. There appears to be a lower trend compared with the chronic myelomonocytic leukemia group, but statistical significance was not reached (P=0.062).
The increased prevalence of CBL gene mutation in chronic myelomonocytic leukemia CMML compared with oligomonocytic chronic myelomonocytic leukemia is intriguing. In particularly, the co-occurrence of CBL along with SRSF2 and TET2 mutations suggests that CBL may collaborate with SRSF2/TET2 in the pathogenesis of chronic myelomonocytic leukemia. CBL is an enzyme catalyzing the transfer of ubiquitin from ubiquitin conjugating enzymes (E2) to substrates, which promotes their degradation by proteasomes. Through this function, CBL serves as a negative regulator for many signal transduction pathways. The absence of CBL mutation in oligomonocytic chronic myelomonocytic leukemia suggests that inactivation of CBL may have an important role in the development of the full phenotype in chronic myelomonocytic leukemia. Thus, while TET2, ASXL1 and SRSF2 represent the driver mutations in both groups, additional mutations, such as CBL, as well as the combinatorial mutation pattern of TET2/ASXL1/SRF2 may influence the final phenotypic characteristics for the two facets of chronic myelomonocytic leukemia (relative monocytosis vs absolute monocytosis, respectively).
TET2, ASXL1 and SRSF2 mutations are also some of the most frequent abnormalities associated with age-related clonal hematopoiesis.26, 27 Gene expression profiling of normal hematopoietic stem cells has revealed an age-related myelomonocytic lineage-bias of elderly stem cells.28, 29 Building on this knowledge, recent whole exome sequencing study of chronic myelomonocytic leukemia patients suggested that chronic myelomonocytic leukemia may arise through successive acquisition of mostly age-related somatic mutations that ultimately convert a myelomonocytic bias into overt leukemia.9 In keeping with these data, at least a subset of oligomonocytic chronic myelomonocytic leukemia may also derive from an accumulation of age-related mutations, and perhaps represents an earlier stage of tumor development, as compared with chronic myelomonocytic leukemia. In this study, the patients in the oligomonocytic group were on average 7 years younger than the patients in the control group. In addition, the percentage of mutations appeared significantly lower in the oligomonocytic compared with conventional chronic myelomonocytic leukemia.
Historically, most of the prognostic tools in chronic myelomonocytic leukemia have been derived from studies focused on myelodysplastic syndrome. Recent sequencing studies noted that ASXL1 mutations, age, hemoglobin, WBC, and platelet counts defined prognostically distinct patient subsets with varied overall survival.12 A ‘CMML-specific prognostic scoring system’ (CPSS) has been proposed based on genetic mutations, cytogenetics and red blood cell transfusion dependence.15, 30 It divides patients into four risk groups with different survival and risk of acute leukemia transformation. Another chronic myelomonocytic leukemia prognostic model from the Mayo Clinic identified absolute monocyte count, presence of circulating immature myeloid cells, anemia and thrombocytopenia as independent variables for survival.20 Thus, even though currently myelodysplastic syndrome and chronic myelomonocytic leukemia patients may have similar therapeutic options, it is likely that future investigation will lead to a more targeted and personalized approach to the treatment of chronic myelomonocytic leukemia.
In conclusion, mutations in ASXL1, TET2 and SRSF2 were frequently found in oligomonocytic chronic myelomonocytic leukemia, indicating the genetic similarities between overt and oligomonocytic chronic myelomonocytic leukemia. These findings suggest that at least a subset of oligomonocytic chronic myelomonocytic leukemia represent early phase ‘dysplastic type’ chronic myelomonocytic leukemia—a fact further substantiated by the development of overt chronic myelomonocytic leukemia in more than one third of oligomonocytic chronic myelomonocytic leukemia cases, possibly through acquisition of additional mutations like CBL. Based on our results, it seems reasonable to classify cases of myeloid neoplasms showing increased peripheral blood monocytes (>0.5 × 109/L), but not in the range of absolute monocytosis required by the current WHO classification as oligomonocytic chronic myelomonocytic leukemia. These patients would be stratified in accordance to chronic myelomonocytic leukemia-specific prognostic scoring systems and may benefit from clinical trial therapies directed at chronic myelomonocytic leukemia. Further studies are required to elucidate any possible disease pathways and the relationship between oligomonocytic and overt chronic myelomonocytic leukemia.
References
Bennett JM, Catovsky D, Daniel MT et al, Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982;51:189–199.
Bennett JM, Catovsky D, Daniel MT et al, Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976;33:451–458.
Vardiman JW, Harris NL, Brunning RD . The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002;100:2292–2302.
Arber DA, Orazi A, Hasserjian R et al, The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–2405.
Geyer JT, Orazi A . Myeloproliferative neoplasms (BCR-ABL1 negative) and myelodysplastic/myeloproliferative neoplasms: current diagnostic principles and upcoming updates. Int J Lab Hematol 2016;38s:12–19.
Kohlmann A, Grossmann V, Klein HU et al, Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 2010;28:3858–3865.
Kosmider O, Gelsi-Boyer V, Ciudad M et al, TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009;94:1676–1681.
Bacher U, Haferlach T, Schnittger S et al, Recent advances in diagnosis, molecular pathology and therapy of chronic myelomonocytic leukaemia. Br J Haematol 2011;153:149–167.
Mason CC, Khorashad JS, Tantravahi SK et al, Age-related mutations and chronic myelomonocytic leukemia. Leukemia 2016;30:906–913.
Meggendorfer M, Roller A, Haferlach T et al, SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012;120:3080–3088.
Yoshida K, Sanada M, Shiraishi Y et al, Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478:64–69.
Itzykson R, Kosmider O, Renneville A et al, Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 2013;31:2428–2436.
Ricci C, Fermo E, Corti S et al, RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res 2010;16:2246–2256.
Palomo L, Garcia O, Arnan M et al, Targeted deep sequencing improves outcome stratification in chronic myelomonocytic leukemia with low risk cytogenetic features. Oncotarget 2016;30:57021–57-35.
Elena C, Galli A, Such E et al, Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood 2016;128:1408–1417.
Orazi A, Germing U . The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia 2008;22:1308–1319.
Lai Z, Markovets A, Ahdesmaki M et al, VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 2016;44:e108.
De Baets G, Van Durme J, Reumers J et al, SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res 2012;40:D935–D939.
Forbes SA, Beare D, Gunasekaran P et al, COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811.
Patnaik MM, Padron E, LaBorde RR et al, Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia 2013;27:1504–1510.
Schuler E, Schroeder M, Neukirchen J et al, Refined medullary blast and white blood cell count based classification of chronic myelomonocytic leukemias. Leuk Res. 2014;38:1413–1419.
Cervera N, Itzykson R, Coppin E et al, Gene mutations differently impact the prognosis of the myelodysplastic and myeloproliferative classes of chronic myelomonocytic leukemia. Am J Hematol 2014;89:604–609.
Mughal TI, Cross NC, Padron E et al, An International MDS/MPN Working Group's perspective and recommendations on molecular pathogenesis, diagnosis and clinical characterization of myelodysplastic/myeloproliferative neoplasms. Haematologica 2015;100:1117–1130.
Wu SJ, Kuo YY, Hou HA et al, The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood 2012;120:3106–3111.
Thol F, Kade S, Schlarmann C et al, Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012;119:3578–3584.
Xie M, Lu C, Wang J et al, Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–1478.
Jaiswal S, Fontanillas P, Flannick J et al, Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–2498.
Pang WW, Price EA, Sahoo D et al, Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA 2011;108:20012–20017.
Beerman I, Bhattacharya D, Zandi S et al, Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA 2010;107:5465–5470.
Such E, Germing U, Malcovati L et al, Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood 2013;121:3005–3015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work was presented in part at the 2015 Annual Meeting of the United States and Canadian Academy of Pathology
Rights and permissions
About this article

Cite this article
Geyer, J., Tam, W., Liu, YC. et al. Oligomonocytic chronic myelomonocytic leukemia (chronic myelomonocytic leukemia without absolute monocytosis) displays a similar clinicopathologic and mutational profile to classical chronic myelomonocytic leukemia. Mod Pathol 30, 1213–1222 (2017). https://doi.org/10.1038/modpathol.2017.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.45
This article is cited by
-
Should we give oligomonocytic chronic myelomonocytic leukemia a higher prominence in the next WHO Classification of Haematolymphoid Tumors?
Leukemia (2023)
-
Advances in myelodysplastic/myeloproliferative neoplasms
Virchows Archiv (2023)
-
Multi-hit TET2 mutations as a differential molecular signature of oligomonocytic and overt chronic myelomonocytic leukemia
Leukemia (2022)
-
Myelodysplastic/myeloproliferative neoplasms-unclassifiable with isolated isochromosome 17q represents a distinct clinico-biologic subset: a multi-institutional collaborative study from the Bone Marrow Pathology Group
Modern Pathology (2022)
-
The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms
Leukemia (2022)
