
Abstract
Chronic lung allograft dysfunction continues to be the main contributor to poor long-term allograft survival after lung transplantation. The restrictive phenotype of chronic lung allograft dysfunction carries a particularly poor prognosis. Little is known about the pathogenetic mechanisms involved in restrictive chronic lung allograft dysfunction. In this study, we performed histomorphological and immunohistochemical analysis of restrictive chronic lung allograft dysfunction lungs. Explant lung tissue from 21 restrictive chronic lung allograft dysfunction patients was collected and histopathologic patterns of rejection, fibrosis and vascular changes were scored after routine histochemical stains and additional immunohistochemistry for endothelial markers and C4d. In all, 75% of cases showed evidence of acute cellular rejection; lymphocytic bronchiolitis was absent in most lungs, whereas in 55% there was obliterative bronchiolitis. Almost half of the cases showed a pattern consistent with pleuroparenchymal fibro-elastosis (n=10), and a subset showed nonspecific interstitial pneumonia (n=5) or irregular emphysema (n=5). Fibrinous alveolar exudates were frequently seen in association with fibrosis (n=6), but no diffuse alveolar damage was found. Evidence of microvascular damage was present in most cases. An emphysematous pattern of fibrosis was associated with a better survival (P=0.0030), whereas fibrinous exudates were associated with a worse survival (P=0.0007). In addition to the previously described nonspecific interstitial pneumonia and pleuroparenchymal fibro-elastosis patterns in restrictive chronic lung allograft dysfunction, we are the first to describe a pattern of fibrosis-induced subpleural/paraseptal emphysema. This pattern confers a better survival, whereas fibrinous exudates are associated with a worse survival. We believe that our findings offer a pathogenetic theory for pleuroparenchymal fibro-elastosis in restrictive chronic lung allograft dysfunction, and show that restrictive chronic lung allograft dysfunction is an increasingly heterogeneous disease with presumably different mechanisms of subpattern formation.
Similar content being viewed by others
Main
Chronic lung allograft dysfunction persists to be one of the main problems limiting long-term survival post lung transplantation, and with a 5-year incidence of 50%, it is one of the major reasons for late mortality and morbidity.1 Two different clinical phenotypes have been distinguished; bronchiolitis obliterans syndrome is defined as an obstructive pulmonary function (forced expiratory volume in 1 second decline >20%, but no decline in forced vital capacity and total lung capacity); restrictive chronic lung allograft dysfunction or restrictive allograft syndrome, on the other hand, is defined as a forced expiratory volume in 1 second decline >20% with a concomitant forced vital capacity decline >20% and/or total lung capacity decline >10%).2 Differential diagnosis remains troublesome with different clinical parameters being proposed and no big cohorts analyzed so far.3 Despite these different diagnostic criteria, it is generally accepted that restrictive chronic lung allograft dysfunction is characterized by persistent infiltrates on computed tomography, whereas bronchiolitis obliterans syndrome more likely shows air trapping/mosaic attenuation.4, 5 Histopathological analysis of bronchiolitis obliterans syndrome lungs shows obliterative bronchiolitis, the typical lesion associated with chronic rejection, being a mostly complete obliteration of the small airway with collagenous matrix, surrounded by a more or less preserved lung parenchyma. In contrast, restrictive chronic lung allograft dysfunction shows a different pattern with severe alveolar fibrosis organized around the pleura and the interlobular septa, remarkably similar to pleuroparenchymal fibro-elastosis, with concomitant obliterative bronchiolitis lesions.6 Pleuroparenchymal fibro-elastosis has previously been recognized as a rare disorder, which is mostly idiopathic and might be associated with frequent infection and auto-immunity,7 but also has been described in the context of allogeneic hematopoietic stem cell transplantation.8 The disease is characterized by mostly apical pleural thickening associated with parenchymal abnormalities, lung distortion and bronchiectasis.9 Next to these findings of pleuroparenchymal fibro-elastosis in lungs of patients suffering from restrictive chronic lung allograft dysfunction, other studies showed that the finding of late onset diffuse alveolar damage predisposed to restrictive chronic lung allograft dysfunction,10, 11 while the study by Paraskeva et al12 demonstrated that patients with acute fibrinous organizing pneumonia show a restrictive pulmonary function and have a very poor survival post-diagnosis.12 Acute fibrinous organizing pneumonia presents with mainly patent bronchioles with a peribronchiolar deposition of intra-alveolar loose fibrillary fibrin filling the alveolar space and minimal presence of inflammatory infiltrates or interstitial thickening. The question, however, is to what degree these findings co-exist and may concur within the same lungs given all these patients present with a clinical situation resembling restrictive chronic lung allograft dysfunction (restrictive pulmonary function, persistent computed tomography infiltrates and poor survival). Especially given the recent report by our own group that there are survival differences across restrictive chronic lung allograft dysfunction patients, with apical-dominated fibrosis being associated with superior survival compared with patients exhibiting basal/diffuse fibrosis.13 Our aim was to identify histopathological alterations present in recipients with a restrictive pulmonary function defect because of chronic lung allograft dysfunction and to relate these histopathological alterations with clinically relevant parameters.
Materials and methods
Patient Selection
We retrospectively reviewed lung tissue of our biobank, collected during autopsy, redo-transplantation or video-assisted thoracoscopic surgery biopsy. The number of blocks available varied (average 3.8, median 3, range 1–11), and was partly dependent on the type of tissue acquisition. Owing to the retrospective nature of the study, exact correlation of computed tomography findings and location of tissue acquired was not possible. Patients with a restrictive pulmonary function defect (either total lung capacity decline >10% and/or forced vital capacity decline with at least 20%) with concomitant persistent computed tomography infiltrates were classified as restrictive chronic lung allograft dysfunction and at least one biopsy randomly taken from the lung, was fixed in formalin, paraffin embedded, sectioned and further used for histopathological analysis. Furthermore, demographic data were assessed, including date of diagnosis, date of redo-transplantation/death, microbiologic cultures/PCR, donor-specific antibody against human leukocyte antibody and data of the last broncho-alveolar lavage before redo-transplantation/death but at/after diagnosis of restrictive chronic lung allograft dysfunction. Broncho-alveolar lavage was performed during indication bronchoscopy (decline in pulmonary function) and was performed with 2 × 50 cc of saline. Both fractions were pooled and sent to the clinical lab for cell differential, microbial and viral analysis via cultures and PCR, respectively.
Analysis
Different histopathologic patterns were scored by an established pathologist experienced in lung transplantation and pleuroparenchymal fibro-elastosis (JHvdT), using hematoxylin–eosin and elastica-van Gieson-stained sections. We scored the presence/absence of specific histopathological patterns (being diffuse alveolar damage/acute fibrinous organizing pneumonia/nonspecific interstitial pneumonia/emphysema/fibro-elastosis/location of fibrosis) with more than one pattern being possible per slice. Fibro-elastosis was distinguished from pre-existing apical caps by the fact that the latter is defined by homogeneous subpleural, collapsed, elastotic tissue, whereas fibro-elastosis is characterized by intra-alveolar aggregates of fibrotic tissue, frequent peribronchial fibrosis and adjacent fibrinous exudates in surrounding alveoli. In addition, patterns of vascular involvement, including capillaritis, vascular obliteration (of capillaries and/or veins) and microvessel density (as confirmed by routine immunohistochemical staining for endothelium with CD34 (clone QBEnd/10) and podoplanin (clone D2-40, to demonstrate lymph vessels)) were analyzed. Moreover, the degree of obliterative bronchiolitis, acute rejection, lymphocytic bronchiolitis and immunohistochemical staining for C4D (clone SP91) were scored according to International Society for Heart and Lung Transplantation guidelines.14 This grading was performed in regions outside of areas of advanced fibrosis. All immunohistochemical staining was performed with ready-to-use antibodies on a Benchmark Ultra system, using an Ultraview Dab kit for visualization (all from Ventana Medical Systems, Oro Vally, AZ, USA).
Statistics
Results are displayed as median (interquartile range) or number (%). Statistics were performed using Mann–Whitney U-test using Prism 6.0. A P-value <0.05 was considered significant.
Results
Lung tissue from 21 patients with restrictive chronic lung allograft dysfunction was analyzed. The first sample was obtained from an autopsy in 2003 and the last in July 2016. The detailed patient characteristics are summarized in Table 1 and histological findings in Table 2. Median survival was 0.59 year (0.28–2.03) after restrictive chronic lung allograft dysfunction diagnosis.
Acute Rejection
Acute rejection was not gradable in five lungs (24%) because of extensive fibrosis, similarly lymphocytic bronchiolitis could not be assessed in seven lungs (33%) because of lack of gradable bronchioles. As for acute rejection, 12/16 (75%) demonstrated evidence of perivascular inflammation, acute rejection was minimal in five patients (A1, 31%), mild in five patients (A2, 31%) and moderate in the remaining two patients (A3, 13%).
Lymphocytic bronchiolitis was absent in most lungs and a maximal grade of B1R was found in three cases only (21%). Of the 21 lungs, 12 showed clear obliterative lesions (55%; Figure 1).

Evidence of acute cellular rejection (a, H&E, Grade 2 rejection), and obliterative bronchiolitis (b, c, H&E and elastica von Gieson stain, respectively). Fibrosis with nonspecific interstitial pneumonia pattern (d, low magnification, e, high magnification), sometimes directly merging into pleuroparenchymal fibro-elastosis pattern (f).
Pattern of Fibrosis
Evaluation of different histologic patterns revealed distinct patterns of fibrosis throughout the different lungs, with often multiple patterns being present within the same biopsy in close proximity to each other. Sharp demarcations with healthy areas could also be observed.
The pleura was fibrotic in all cases where pleura was evaluable on biopsy (n=20), this varied from mild (n=5, 25%), to moderate (n=11, 55%), to severe (n=4, 20%) fibrosis.
Almost half of the cases showed a pattern of intra-alveolar fibrosis consistent with pleuroparenchymal fibro-elastosis (n=10, 48%), whereas a slightly lower number demonstrated organizing pneumonia (n=9, 43%). Other common findings included nonspecific interstitial pneumonia (n=5, 24%, in one case this was the sole pattern of fibrosis; Figure 1) and bland fibrosis with emphysema, which was interpreted as fibrosis-induced subpleural/paraseptal emphysema (n=5, 24%).
Secondary Histological Patterns
Presence of fibrinous exudates was frequently seen on the border of pleuroparenchymal fibro-elastosis and adjacent structurally normal lung, ranging from pure fibrin to a pattern of acute fibrinous organizing pneumonia (n=6, 29%), whereas in some cases, this pattern of fibrinous filling of alveolar air spaces was more diffuse (Figure 2). Focal obliteration of blood vessels was seen in a majority of cases (n=18, 86%), and high-power morphological analysis revealed that these fibrinous exudates corresponded to areas of microvascular damage, obliteration and thrombotic occlusion. The morphology was suggestive of possible antibody-mediated rejection with capillaritis in four cases, despite a lack of convincing C4d staining. Areas of fibrin deposition merged directly into intra-alveolar fibrosis in a subpleural pattern in most cases, and into diffuse intra-alveolar fibrosis and peribronchiolar/paraseptal intra-alveolar fibrosis in a minority of cases (Figure 3). Focal decrease in microvascular density in the form of complete capillary obliteration was corroborated by immunohistochemical staining for endothelial cells (CD34), with exuberant reactive microvascular proliferation in areas of fibrosis. Ectasia of small lymphatic vessels was also frequently seen in parenchyma adjacent to areas of pleuroparenchymal fibro-elastosis (n=6), and staining for D2-40 showed aberrant expression in alveolar septa in some cases (Figure 4).

Patterns of intra-alveolar fibrin deposition, ranging from pure fibrin exudates (a, H&E), to organizing pneumonia (b, H&E) and acute fibrinous organizing pneumonia (c, H&E stain). (d) Evidence of capillaritis suggestive of humoral rejection (H&E stain). (e, f) Damage of intraseptal capillaries with associated fibrin exudation (H&E, low- and high-power magnification, respectively).

Patterns of progression of intra-alveolar fibrin to fibrosis (H&E and elastica von Gieson stains, respectively), with subpleural (a+b), diffuse (c+d) and peribronchial patterns (e+f).

(a) Ectatic septal capillaries (H&E). (b+c). Obliteration of capillaries in areas of ongoing fibrosis (CD34 stain, subpleural and diffuse, respectively). (d) Lymphatic congestion. (e+f). Aberrant lymphatic differentiation and proliferation, respectively, (D2-40 stain).
Association of Histological Pattern and Survival
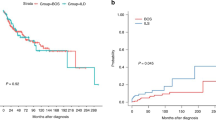
Sub-dividing our patient cohort in patients with a short post-diagnosis graft survival (either death or redo-transplantation) and a better survival post diagnosis utilizing a threshold of the median survival (0.59 year), we observe that acute fibrinous organizing pneumonia was a unique finding in those patients with a survival time <0.59 year, whereas pleuroparenchymal fibro-elastosis was also frequent (n=5, although in three only the onset of pleuroparenchymal fibro-elastosis could be seen; there were no mature lesions), whereas organizing pneumonia (n=6) and nonspecific interstitial pneumonia (n=2) are also found, but emphysema was completely absent. In the patients with a post-diagnosis survival ≥0.59 year, the main observation is pleuroparenchymal fibro-elastosis (n=5), whereas nonspecific interstitial pneumonia (n=3) and organizing pneumonia (n=3) are also frequently found. Remarkably, five of these lungs show fibrosis-induced subpleural/paraseptal emphysema. In fact, all of the lungs with a post-restrictive chronic lung allograft dysfunction diagnosis survival time over 3 years (n=4) showed some degree of emphysema. Overall survival analysis revealed that an emphysematous pattern of fibrosis was associated with a significantly higher chance of survival when compared with the other patterns (P=0.0030, Figure 5). Similarly, a fibrinous pattern was associated with a significantly worse survival (P=0.0007, Figure 5).

(a) Kaplan–Meier curves comparing survival of acute fibrinous organizing pneumonia and emphysema type patterns of fibrosis in restrictive chronic lung allograft dysfunction patients. (b) Representative section of extensive fibrosis-induced paraseptal and subpleural emphysema, with a predominant centrilobular pattern (secondary pulmonary lobule indicated by gray line; asterisk denotes relatively normal parenchyma in adjoining lobule; H&E stain, magnification × 1.25).
Association of Histological Pattern and Broncho-Alveolar Lavage
Broncho-alveolar lavage at restrictive chronic lung allograft dysfunction diagnosis was available in 18 patients (86%, broncho-alveolar lavage not performed because previous bronchiolitis obliterans syndrome, n=3). More details including total and percentage differential cell counts can be found in Table 1. The histological pattern of pleuroparenchymal fibro-elastosis and organizing pneumonia were not associated with any of the broncho-alveolar lavage cell counts. The finding of acute fibrinous organizing pneumonia was associated with increased % and total cell counts of eosinophils in broncho-alveolar lavage at diagnosis (P=0.035 and P=0.013, respectively), but not with any of the other parameters. Emphysema was inversely associated with a lower total cell count (P=0.0050) and the absolute neutrophil count (P=0.0063).
Association with Other Factors
There were no associations between specific histological patterns and time from transplantation to restrictive chronic lung allograft dysfunction diagnosis for any of the parameters studied (acute fibrinous organizing pneumonia, P=0.94; pleuroparenchymal fibro-elastosis, P=0.37). Underlying disease and age was not associated with specific histological patterns; specifically, there was no association with the underlying disease pattern in the five patients who had been transplanted for interstitial lung disease (these included three sarcoidosis, one idiopathic pulmonary fibrosis and one systemic sclerosis case) There was also no association between the location of the infiltrates on computed tomography (upper vs lower vs diffuse) and typical histological patterns; for example, within the acute fibrinous organizing pneumonia group there was one patient with apical infiltrates, whereas in the non-acute fibrinous organizing pneumonia group 6/15 patients showed apical fibrosis (P=0.33). Similarly, in the pleuroparenchymal fibro-elastosis group there were 4/10 (40%) with apical dominant infiltrates on computed tomography, whereas in the non-pleuroparenchymal fibro-elastosis group there were 3/11 (27%).
Discussion
The pattern of pleuroparenchymal fibro-elastosis has previously been described as the primary type of fibrosis associated with to restrictive chronic lung allograft dysfunction, which was also corroborated in our series. In addition, we found nonspecific interstitial pneumonia and fibrosis-induced subpleural/paraseptal emphysema as histopathological patterns in a substantial number of patients. Remarkably, acute fibrinous organizing pneumonia seemed to be a unique findings in patients with worse post-diagnosis outcome, whereas emphysema was associated with a better survival. In the previous case series, emphysema has not been described as histological pattern in restrictive chronic lung allograft dysfunction lungs, and pleuroparenchymal fibro-elastosis and nonspecific interstitial pneumonia were rather common findings. In the context of lung graft-versus-host disease post hematopoietic stem cell transplantation, nonspecific interstitial pneumonia has been found in 75% of the inspected lungs, of which most showed fibrotic nonspecific interstitial pneumonia and the remainder a pattern of cellular nonspecific interstitial pneumonia.15 The underlying etiology of nonspecific interstitial pneumonia-type fibrosis in the context of transplantation has not been resolved, but may include (a combination of) immune phenomena and side effects of medication. We found a lower abundance of nonspecific interstitial pneumonia in our lungs; however, this in agreement with the report by Ofek et al6 who described nonspecific interstitial pneumonia in 25% of their patients.
Remarkably, the typical diffuse alveolar damage pattern with hyaline membrane formation was not observed in any of the lungs, which is in sharp contrast with the previous histological description of restrictive chronic lung allograft dysfunction.10, 11 However, fibrin deposition was a frequently observed finding, often in the context of acute fibrinous organizing pneumonia, which is in concordance with the recent findings of Jonigk et al.16 It is possible that the time between the initial injury and death/re-transplantation was too long to observe the typical initial pattern of diffuse alveolar damage. Alternatively, diffuse alveolar damage might not actually be causally related to pleuroparenchymal fibro-elastosis, and while the article by Ofek et al described this pattern,6 this preceded the article by Paraskeva et al,12 which was the first report on acute fibrinous organizing pneumonia in lung transplant recipients, and this might explain why this specific pattern was not described. Thus, it is possible that some of the diffuse alveolar damage described in the literature might in fact be non-hyaline fibrin deposition more akin to that described herein and by Jonigk et al.16
As for the cause of fibrin deposition, the study by Jonigk et al does not offer an explanation. In our study, however, we found evidence for microvascular damage, which overlapped with areas of fibrin presence in alveoli, be it in in a localized or more diffuse pattern. This included (lymphatic) ectasia, capillaritis, microvascular thrombi, physical breaks in the vascular wall with associated fibrin exudation, luminal obliteration and aberrant microvascular proliferation in areas of fibrosis. Although our observations lend support to the hypothesis that microvascular injury may be pivotal in the cascade of fibrin deposition and ensuing intra-alveolar fibrosis, the exact nature of the microvascular damage remains to be elucidated. In the context of chronic lung allograft dysfunction, this may well be, at least partially, because of chronic (subclinical) humoral rejection, which remains a histologically poorly defined entity in lung transplant pathology. However, only 35% of patients had donor-specific human leukocyte antigen antibodies. Therefore, the exact nature of the microvascular injury remains to be investigated. The sequelae of this fibrin accumulation are likely to correspond to the sequence of events described by Jonigk et al, although we did not find morphological evidence of an intervening phase of macrophage accumulation. On the contrary, in many of our cases, areas of fibrin appear to merge directly with fibrosis of the pleuroparenchymal fibro-elastosis type. Thus, it is likely that acute fibrinous organizing pneumonia is one of the early phases of the disease processes with fibrin being deposited because of (microvascular) injury, possibly in part because of antibody-mediated rejection. Incomplete resolution because of defective clearance of this material or another (potentially) new stimulus, may then evolve to a pleuroparenchymal fibro-elastosis pattern with the typical (sub)pleural, peribronchiolar and paraseptal fibrotic alveolar filling, resulting in the typical restrictive pulmonary function of the patient. This might also explain the step-wise pattern of decline of these patients with each new injury resulting in a new phase with disease progression.17 Although we have to acknowledge that we cannot demonstrate this sequence conclusively, as we have worked with (mostly) end-stage lung biopsies and the post-chronic lung allograft dysfunction diagnosis survival in some of our cases is very limited.
Clinically, it is also of interest that acute fibrinous organizing pneumonia was uniquely present in those patients with a worse survival after restrictive chronic lung allograft dysfunction diagnosis, whereas emphysema was associated with a better survival. The areas of increased tissue fibrosis likely increase traction on the surrounding lung tissue, which might eventually result in an emphysema-like pattern, which is seen in the lungs with the longest post- restrictive chronic lung allograft dysfunction survival. It is of interest that those lungs with an emphysema-like pattern show reduced inflammation in broncho-alveolar lavage at the moment of diagnosis.
Although with respect to evidence of rejection, we found a relatively low incidence of obliterative bronchiolitis lesions, in contrast to the two previous articles,6, 18 this could be due to sample bias19 and more (central) slices would probably reveal obliterative bronchiolitis lesions in all the investigated lungs. Evidence of acute cellular rejection, as found in a substantial number of the patients in our series, on the other hand, is a novel histopathological pattern in restrictive chronic lung allograft dysfunction, and has previously even been regarded as an exclusion criterion for the diagnosis of chronic lung allograft dysfunction. As we, however, believe that chronic vascular rejection may well have a role in the development of fibrotic lung disease following lung transplantation, this does not appear to be an incongruous finding. Moreover, both these findings are in line with a previous similar case series description where acute rejection was present in all investigated lungs and where a variable degree of obliterative bronchiolitis was observed.20 In general, scoring of bronchioles in our study was hampered by the fact that because of the often advanced nature of the abnormalities, areas of fibrotic consolidation were found, incorporating pre-existing bronchioles and making assessment of individual obliterative and lymphocytic bronchiolitis lesions difficult.
In summary, we believe that we have made considerable progress in defining the detailed histomorphological spectrum of chronic lung allograft dysfunction and understanding its possible etiology and pathogenic mechanisms. In addition, we have found distinct patterns with differential prognostic implications in this patient population, and in concert with the detailed description of histological changes provided by this substantial series, we are confident that this will provide a good basis for future mechanistic research and the development of clinical approaches for the treatment of this debilitating disease that continues to mar the long-term outcome of lung transplantation.
References
Yusen RD, Edwards LB, Kucheryavaya AY et al. The registry of the International Society for Heart and Lung Transplantation: Thirty-Second Official Adult Lung and Heart-Lung Transplantation Report-2015; focus theme: early graft failure. J Heart Lung Transplant 2015;34:1264–1277.
Meyer KC, Raghu G, Verleden GM et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
Verleden SE, Ruttens D, Vandermeulen E et al. Restrictive chronic lung allograft dysfunction: where are we now? J Heart Lung Transplant 2015;34:625–630.
Todd JL, Jain R, Pavlisko EN et al. Impact of forced vital capacity loss on survival after the onset of chronic lung allograft dysfunction. Am J Respir Crit Care Med 2014;189:159–166.
Sato M, Waddell TK, Wagnetz U et al. Restrictive allograft syndrome (RAS): a novel form of chronic lung allograft dysfunction. J Heart Lung Transplant 2011;30:735–742.
Ofek E, Sato M, Saito T et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod Pathol 2013;26:350–356.
Reddy TL, Tominaga M, Hansell DM et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J 2012;40:377–385.
von der Thüsen JH, Hansell DM, Tominaga M et al. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol 2011;24:1633–1639.
von der Thüsen JH . Pleuroparenchymal fibroelastosis: its pathological characteristics. Curr Respir Med Rev 2013;9:238–247.
Sato M, Hwang DM, Ohmori-Matsuda K et al. Revisiting the pathologic finding of diffuse alveolar damage after lung transplantation. J Heart Lung Transplant 2012;31:354–363.
Shino MY, Weigt SS, Li N et al. CXCR3 ligands are associated with the continuum of diffuse alveolar damage to chronic lung allograft dysfunction. Am J Respir Crit Care Med 2013;188:1117–1125.
Paraskeva M, McLean C, Ellis S et al. Acute fibrinoid organizing pneumonia after lung transplantation. Am J Respir Crit Care Med 2013;187:1360–1368.
Verleden SE, Ruttens D, Vandermeulen E et al. Predictors of survival in restrictive chronic lung allograft dysfunction after lung transplantation. J Heart Lung Transplant 2016;35:1078–1084.
Stewart S, Fishbein MC, Snell GI et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant 2007;26:1229–1242.
Takeuchi Y, Miyagawa-Hayashino A, Chen F et al. Pleuroparenchymal fibroelastosis and non-specific interstitial pneumonia: frequent pulmonary sequelae of haematopoietic stem cell transplantation. Histopathology 2015;66:536–544.
Jonigk D, Rath B, Borchert P et al. Comparative analysis of morphological and molecular motifs in bronchiolitis obliterans and alveolar fibroelastosis after lung and stem cell transplantation. J Pathol Clin Res 2017;3:17–28.
Sato M, Hwang DM, Waddell TK et al. Progression pattern of restrictive allograft syndrome after lung transplantation. J Heart Lung Transplant 2013;32:23–30.
Verleden SE, Vasilescu DM, McDonough JE et al. Linking clinical phenotypes of chronic lung allograft dysfunction to changes in lung structure. Eur Respir J 2015;46:1430–1439.
Verleden SE, Vasilescu DM, Willems S et al. The site and nature of airway obstruction after lung transplantation. Am J Respir Crit Care Med 2014;189:292–300.
Martinu T, Howell DN, Davis RD et al. Pathologic correlates of bronchiolitis obliterans syndrome in pulmonary retransplant recipients. Chest 2006;129:1016–1023.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
von der Thüsen, J., Vandermeulen, E., Vos, R. et al. The histomorphological spectrum of restrictive chronic lung allograft dysfunction and implications for prognosis. Mod Pathol 31, 780–790 (2018). https://doi.org/10.1038/modpathol.2017.180
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.180
This article is cited by
-
Intragraft immune cells: accomplices or antagonists of recipient-derived macrophages in allograft fibrosis?
Cellular and Molecular Life Sciences (2023)
-
Pleuroparenchymal fibroelastosis after hematopoietic stem cell transplantation in children: a propensity score–matched analysis
European Radiology (2022)
-
Phenotypical diversity of airway morphology in chronic lung graft vs. host disease after stem cell transplantation
Modern Pathology (2019)
