
Abstract
Infantile fibrosarcoma and congenital mesoblastic nephroma are tumors of infancy traditionally associated with the ETV6–NTRK3 gene fusion. However, a number of case reports have identified variant fusions in these tumors. In order to assess the frequency of variant NTRK3 fusions, and in particular whether the recently identified EML4–NTRK3 fusion is recurrent, 63 archival cases of infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma and secretory breast carcinoma (tumor types that are known to carry recurrent ETV6–NTRK3 fusions) were tested with NTRK3 break-apart FISH, EML4–NTRK3 dual fusion FISH, and targeted RNA sequencing. The EML4–NTRK3 fusion was identified in two cases of infantile fibrosarcoma (one of which was previously described), and in one case of congenital mesoblastic nephroma, demonstrating that the EML4–NTRK3 fusion is a recurrent genetic event in these related tumors. The growing spectrum of gene fusions associated with infantile fibrosarcoma and congenital mesoblastic nephroma along with the recent availability of targeted therapies directed toward inhibition of NTRK signaling argue for alternate testing strategies beyond ETV6 break-apart FISH. The use of either NTRK3 FISH or next-generation sequencing will expand the number of cases in which an oncogenic fusion is identified and facilitate optimal diagnosis and treatment for patients.
Similar content being viewed by others
Main
Infantile fibrosarcoma (also known as congenital fibrosarcoma) is a malignant tumor of infancy.1 The classic presentation is a rapidly enlarging mass on the extremity of a child under 2 years of age. Most cases of infantile fibrosarcoma are diagnosed during the first year of life, although tumors have occasionally been detected in utero.2, 3 The histopathology varies greatly, and the differential diagnosis is broad. The classic form is composed of long fascicles of densely cellular spindle cells with a variable amount of collagenous stroma. On the other side of the morphologic spectrum, infantile fibrosarcoma can be organoid, with a hemangiopericytoma-like vascular pattern, and plump cells with ovoid nuclei, more rounded cytoplasm, and scant poorly collagenous stroma. This latter appearance may be indistinguishable from infantile hemangiopericytoma, a distinct morphologic variant of infantile myofibroma/myofibromatosis. Mitoses, apoptosis and areas of necrosis are frequently present in infantile fibrosarcoma, but again, these features are variable and zonal. Not uncommonly, it has a mononuclear inflammatory infiltrate scattered throughout the tumor. The clinical outcome is generally favorable, with rare metastases and a mortality rate of <5%, although tumors recur in up to 50% of patients and patients typically require a combination of chemotherapy and surgery.1, 3, 4, 5, 6, 7, 8, 9 ETV6–NTRK3 translocations are recurrent in infantile fibrosarcoma, reported in approximately 70% of cases.10 This rearrangement results in a fusion protein between NTRK3 (also known as NT-3 growth factor receptor or TrkC) and the transcription factor ETV6 with resultant upregulation of the Ras-MAPK and PI3K-Akt pathways.11, 12, 13, 14, 15 Molecular techniques to identify this fusion are a component of the standard diagnostic work-up for these tumors.
Congenital mesoblastic nephroma is a renal tumor of infancy that, like infantile fibrosarcoma, is typically diagnosed within the first year of life. Surgical resection is adequate treatment in most cases, with a 5-year survival of ~95%.16 Based on histologic appearance, congenital mesoblastic nephromas are divided into classic, cellular and mixed types, with the cellular type being histologically identical to infantile fibrosarcoma. Like infantile fibrosarcoma, cellular congenital mesoblastic nephroma typically harbors the ETV6–NTRK3 rearrangement.17, 18 The ETV6–NTRK3 fusion has also been shown to be oncogenic in multiple additional tumor types, including mammary analog secretory carcinoma, secretory breast carcinoma and papillary thyroid carcinoma.19, 20, 21, 22, 23, 24
At the time of diagnosis, suspected cases of infantile fibrosarcoma and congenital mesoblastic nephroma are routinely assessed for the ETV6–NTRK3 rearrangement in order to support the histopathologic diagnosis. ETV6 break-apart fluorescence in situ hybridization (FISH) or reverse transcriptase-PCR (RT-PCR) specific to the ETV6–NTRK3 fusion are the most common tests used to screen for this rearrangement.11, 21, 22, 25, 26, 27 These techniques are necessary as the t(12;15) associated with ETV6–NTRK3 fusion is not always visible on karyotype, and karyotype may not always be successful or available. NTRK3 immunohistochemical staining is generally not reliable in detecting ETV6–NTRK3 rearranged tumors.14, 15
ETV6 break-apart FISH is negative in approximately 30% of infantile fibrosarcoma cases, however, suggesting the possibility of alternate translocations in this tumor type.13 Recently, case reports of novel translocations have been described in infantile fibrosarcoma, including EML4–NTRK3 and LMNA–NTRK1,28, 29 whereas cases of ETV6 fused to unknown gene partners, referred to as ETV6-X, have been reported in mammary analog secretory carcinoma.30, 31 In the past, such novel fusions may have been of more academic than practical interest. Today, however, their identification carries significant therapeutic implications with recent reports describing treatment of infantile fibrosarcoma with targeted therapy. One report describes treating a patient with infantile fibrosarcoma with a targeted inhibitor of the tropomyosin-related kinase (TRK) family, of which NTRK3 is one member, as part of a Phase 1 trial of the drug.32 The completed trial showed a 91% objective response rate to larotrectinib in pediatric patients with solid tumors harboring confirmed NTRK1, NTRK2 or NTRK3 fusions.33 Another report describes treating a patient with infantile fibrosarcoma with the novel LMNA–NTRK1 fusion with crizotinib, which has been shown to have activity against both NTRK1-and NTRK3-dependent tumors.29, 34 Both reported patients showed significant responses to these targeted therapies. An additional recent article describes a patient with secretory breast carcinoma and an ETV6–NTRK3 fusion who also responded to targeted TRK inhibition.35
In order to treat patients with targeted therapies, however, the presence of the target must be confirmed, yet the current standard assays (ETV6 break-apart FISH or ETV6–NTRK3 RT-PCR) will miss cases with alternative fusion partners. In order to establish whether the EML4–NTRK3 fusion is recurrent, and to evaluate testing strategies that would capture alternative fusions, we collected archival cases whose differential diagnosis included infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma or secretory breast carcinoma, and screened these cases for rearrangements using either novel FISH probes, targeted multiplex sequencing, or both.
Materials and methods
Case Selection
Sixty-three archival cases from 63 patients of known or suspected infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma or secretory breast carcinoma were collected from eight institutions. The original surgeries occurred between 1980 and 2015. Case demographics are detailed in Table 1. Note that one case was previously reported.28
Fluorescence In Situ Hybridization
NTRK3 break-apart FISH
Custom FISH probes flanking the NTRK3 gene were designed with Agilent SureFISH software. The green probe set, comprised of 46 610 oligos spans a region of 400.295 kbp around the 5′ end of NTRK3. The orange/red probe set, comprised of 42 417 oligos, spans a region of 398.66 kbp around the 3′ end of NTRK3.
EML4–NTRK3 dual fusion FISH
Custom FISH probes were designed to specifically detect the EML4–NTRK3 fusion (Agilent SureFISH). The green probe set, comprised of 131 044 oligos spans a region of 1.363 Mbp around the 5′ end of EML4. The orange/red probe set, comprised of 165 307 oligos spans a region of 1.597 Mbp around the 3′ end of NTRK3.
FISH probes were hybridized to metaphase cells before use in these experiments in order to confirm hybridization to the correct chromosome.
Formalin-fixed paraffin-embedded tissue slides cut at 5 μm thickness were used, prepared according to standard laboratory practices at their institutions of origin. Probes were hybridized according to standard protocols using DAKO Histology FISH accessory kits. Fifty nuclei were scored using an orange and green dual filter set and individual orange and green filters as needed. Images were captured using the Applied Imaging CytoVision Imaging System. Interpretation cutoffs were determined independently for each probe set by the beta inverse calculation.
High-Throughput Sequencing for Fusion Detection
Forty-four cases were tested with a targeted high-throughput sequencing assay, using an RNA-based anchored multiplex PCR library preparation method (ArcherDX). Nucleic acids were extracted from formalin-fixed paraffin-embedded tissue on unstained slides using Agencourt FormaPure (Beckman Coulter, cat # A33341) according to the manufacturer’s total nucleic acid purification protocol. RNA was quantified with Qubit RNA HS Assay Kit (ThermoScientific, cat # Q32852) after purification. Functional RNA quality was assessed in-line with library generation.
Libraries were prepared according to Archer FusionPlex Comprehensive Thyroid and Lung Panel (ArcherDX, cat # SK0070) Protocol Revision C. 200 ng RNA—quantified by Qubit—was used as input for library generation with Archer Universal RNA Reagent Kit v2 (ArcherDX, cat # AK0040-8), Archer™ MBC Adapters Set A for Illumina® (ArcherDX, cat # SA0040) and the Archer FusionPlex Sarcoma Panel (ArcherDX, cat # AK0032-8). Gene targets include selected exons of: ALK, CAMTA1, CCNB3, CIC, EPC, EWSR1, FKHR, FUS, GLI1, HMGA2, JAZF1, MEAF6, MKL2, NCOA2, NTRK3, PDGFB, PLAG1, ROS1, SS18, STAT6, TAF15, TCF12, TFE3, TFG, USP6 and YWHAE. All purifications during library preparation were performed with Agencourt AMPure XP (Beckman Coulter, cat # A63882).
RNA quality was assessed in-line with library generation by the Archer PreSeq RNA QC Assay (ArcherDX, cat #AK0043-16), provided with the Universal Reagent Kit, according to kit protocol. Cycling and data collection was performed on a StepOne™ Real-Time PCR System (ThermoScientific, cat # 4376357). The default threshold of 0.078 was used for all samples.
Final libraries were quantified using the KAPA Library Quantification Kit (KAPA Biosystems, cat # KK4824) and pooled to equimolar concentration. The intended concentration of pooled libraries was confirmed with the KAPA Library Quantification Kit.
Sequencing
Libraries were sequenced on an Illumina NextSeq 500 using NextSeq 500 v2 reagents (Illumina, cat # FC-404-2005) for paired-end, 150 base pair reads and dual index reads. Samples were multiplexed such that each library was sequenced to at least 2 million paired reads. The flow cell was loaded with 1.8 pM denatured library and 20% PhiX Control v3 (Illumina, cat # FC-110-3001).
Data Analysis
All sequences were recorded in GRCH37 (hg19) coordinates. Data were analyzed by Archer Analysis version 4.0. Briefly, adapter sequences were trimmed from the reads, then PCR duplicates were collapsed using molecular barcodes to identify unique molecules. Split alignments indicated a putative fusion event and a consensus sequence was built from the unique molecules supporting a given fusion event. The consensus was BLASTed to the reference genome and fusion annotation was guided by RefSeq. Finally, a set of filters was applied to the annotated result to reduce false positives and characterize a fusion event as known or novel. Interpretation of the fusion sequence was made by a board-certified molecular genetic pathologist.
Results
In order to establish whether EML4–NTRK3 fusions are recurrent in tumor types known to have recurrent ETV6–NTRK3 fusions, as well as to evaluate testing strategies that would capture alternative fusions, we conducted a screen of archival cases with a differential diagnosis that included infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma or secretory breast carcinoma. Sixty-three cases of known or suspected infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma and secretory breast carcinoma were collected from eight institutions. Many of these cases had undergone prior testing for an ETV6–NTRK3 translocation, typically using either ETV6 break-apart FISH or ETV6–NTRK3 RT-PCR. Karyotypes were also available for a subset of cases, which may be highly suggestive of an ETV6–NTRK3 translocation in the appropriate clinical context (ie, the finding of a t(12;15)(p13;q25) in a tumor with histology consistent with IFS). Table 1 provides details of original diagnosis and prior testing.
Prior targeted testing for an ETV6–NTRK3 translocation had been performed in 23 of 63 (37%) cases. In addition, 26 (41%) of cases had karyotype testing (without targeted testing). Of the cases with a positive finding of ETV6 translocation (with either FISH or RT-PCR), 100% were given a final pathologic diagnosis of the initially suspected tumor type (infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma or secretory breast carcinoma), whereas of the negative cases, the final pathologic diagnosis was descriptive (eg, ‘high-grade sarcoma’) in 7 of 13 (54%), demonstrating the significance of molecular findings to the diagnostic process.
In order to identify all cases of NTRK3 fusion in this group of cases, regardless of fusion partner, an NTRK3 break-apart probe set was created. Interphase FISH using this probe set was performed on 59 cases and was technically successful in all but one (98%). In all, 34 of 58 cases (59%) were positive for an NTRK3 rearrangement, including 25 of 26 (96%) of infantile fibrosarcoma, 7 of 18 (39%) of congenital mesoblastic nephroma, 3 of 3 (100%) of mammary analog secretory carcinoma and 2 of 3 (67%) of secretory breast carcinoma. Eight cases with other diagnoses were negative for NTRK3 rearrangement. All cases that were previously identified to have an ETV6 rearrangement by either FISH or RT-PCR were positive for an NTRK3 rearrangement using the novel probe set, confirming its ability to detect the canonical ETV6–NTRK3 rearrangement.
Of the three cases with NTRK3 rearrangement but negative for ETV6 rearrangement by original testing, one infantile fibrosarcoma case was known to carry an EML4–NTRK3 rearrangement from prior work (case 58).28 Therefore, a novel EML4–NTRK3 dual fusion FISH probe set was created and 46 cases with sufficient material were tested to determine whether EML4 is a recurrent alternate translocation partner in tumors known to carry common ETV6–NTRK3 fusions. FISH using this probe set was technically successful in 45 out of 46 cases. The case previously known to carry the EML4–NTRK3 fusion was positive using this novel assay, as were two additional cases: a second case of infantile fibrosarcoma and a case of congenital mesoblastic nephroma. The histology in these cases was typical of that seen in ETV6–NTRK3 rearranged infantile fibrosarcoma and congenital mesoblastic nephroma (Figure 1). No cases with a previously detected ETV6–NTRK3 translocation were positive by this assay.

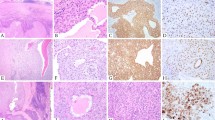
Three cases with the EML4–NTRK3 fusion are identified. Case 3, infantile fibrosarcoma: large abdominal mass composed of medium-sized plump oval cells arranged in broad bands separated by thin-walled, ectatic blood vessels. Nuclei are ovoid, mildly pleomorphic with open chromatin and an inconspicuous single nucleolus. Tumor cells undergoing mitoses and apoptosis are frequently observed. Case 58, infantile fibrosarcoma: cellular spindle cell sarcoma characterized by long intersecting fascicles of uniform spindle cells with scant collagenous stroma. Nuclei are elongated with fine chromatin and inconspicuous nucleoli. Mitoses are frequently observed. Case 41, congenital mesoblastic nephroma: cellular spindle cell tumor vaguely arranged in short fascicles. Tumor cells are uniform with round to ovoid to elongated nuclei with open chromatin and inconspicuous nucleoli. Scant collagenous stroma and frequent mitoses are observed. The histology (a, e, i), positive NTRK3 break-apart FISH results (b, f, j), positive EML4–NTRK3 dual fusion FISH results (c, g, k) and fusion sequence reads (d, h) are shown for each case. Note that the sequencing assay for case 41 failed quality control metrics and did not return a result. The case was tested 20 years after specimen collection.
A subset of the collected cases still did not have an identified oncogenic fusion after testing by FISH, therefore 44 cases with sufficient tissue were prepared to run on a targeted RNA-based next-generation sequencing fusion detection panel (ArcherDX). Twenty-three of these cases passed pre-sequencing quality control thresholds. The likelihood that a case would fail quality control increased with the age of the sample, which is an expected result for an assay using RNA extracted from formalin-fixed paraffin-embedded tissue. Of cases <5 years old at the time of RNA extraction, 95% (19 out of 20) passed quality control. Of the 23 total cases that passed quality control, 16 fusions were identified (Table 1), all of which corresponded to the chromosomal alterations identified by FISH in the current work, or by FISH, karyotype, and/or RT-PCR in previous testing, including 14 cases with ETV6–NTRK3 translocation and 2 cases with EML4–NTRK3 translocation. In addition, 21 cases did not pass pre-sequencing quality control but were sequenced anyway. Despite the poor quality scores, 7 of the 21 had fusions identified, and 6 of 7 corresponded to positive FISH results. Five cases with poor sequencing quality scores had rearrangements by FISH that were not identified by sequencing. On average, these were older cases (7 to 23 years, mean 13 years, s.d. 6.4 years from the date of collection to the date of nucleic acid extraction).
In total, of the 44 tumors sequenced, 21 ETV6–NTRK3 fusions were identified, and 2 EML4–NTRK3 fusions. Each of the ETV6–NTRK3 fusions identified with this analysis connects ETV6 exon 5 (NM_001987.4) to NTRK3 exon 15 (NM_001243101.1). Both of the EML4–NTRK3 fusions identified with this analysis connect EML4 exon 2 (NM_019063.4) to NTRK3 exon 14 (NM_001243101.1).
Overall, the results of the different targeted assays (FISH and/or sequencing) showed good concordance with each other. For cases tested by NTRK3 break-apart FISH, where another targeted method was also performed, 17 of 19 positive cases, and 12 of 12 negative cases correlated with the other result; cases 57 and 62 had conflicting results (Table 1). For cases tested by EML4–NTRK3 dual fusion FISH, where another targeted method was available to confirm the result, all results correlated (3 of 3 positive and 34 of 34 negative). For cases tested by sequencing that passed quality control, 15 of 16 positive cases and 7 of 7 negative cases correlated with other results; case 57 had conflicting results, as mentioned above (Table 1). Both of the discordant cases had ETV6 break-apart FISH that was negative at the time of initial work-up, with positive NTRK3 break-apart FISH and ETV6–NTRK3 sequencing results in the current work.
Discussion
Here we describe the recurrent nature of the novel EML4–NTRK3 fusion in infantile fibrosarcoma and congenital mesoblastic nephroma, now detected in two additional cases beyond the index case.28 The fusions include exons 1 and 2 of EML4 and exons 15 through 20 of NTRK3. These breakpoints are similar to those that have been reported in other EML4- and NTRK3 rearranged tumors. The EML4–ALK translocation is recurrently described in non-small cell lung carcinoma.36 Breakpoints in EML4 are variable in these tumors, but always preserve the coiled-coil domain of the EML4 protein (echinoderm microtubule-associated protein-like 4), encoded by exon 2 (NM_019063.4).37 The chimeric protein shows constitutive dimerization with resultant activation of the tyrosine kinase domain of ALK.36 The classic infantile fibrosarcoma and congenital mesoblastic nephroma translocation ETV6–NTRK3 has a similar functional profile, with preservation of the helix loop helix domain of ETV6 and the kinase domain of NTRK3 resulting in oligomerization of the fusion protein and constitutive activation of the NTRK3 tyrosine kinase. Although the EML4-NTRK3 translocation is novel, the EML4 and NTRK3 breakpoints observed in the current analysis are similar to those that have been previously reported in EML4-ALK and ETV6–NTRK3 fusions, respectively, and the resulting fusion protein would be predicted to include the coiled-coil domain of EML4 and the kinase domain of NTRK3 (Figure 2). The EML4–NTRK3 fusion has been shown to be tumorigenic in vitro and in vivo.28

The ETV6–NTRK3 and EML4–NTRK3 fusion products are similar, with the 5′ partner contributing the binding domain with conservation of the NTRK3 kinase domain in the 3′ position.
Combining these data with previous descriptions of alternate rearrangements in infantile fibrosarcoma and related cancers challenges, the notion that these tumors are defined by the ETV6–NTRK3 translocation alone. Alternative translocations are increasingly described in cancer,38 so it may come as no surprise that this group of tumors is similarly diverse. Importantly, the diversity and recurrent nature of these novel translocations, as well as their potential therapeutic significance, requires a reassessment of the standard clinical practice of using ETV6 FISH or ETV6–NTRK3 RT-PCR to identify fusions. These technologies are robust, but limited in their scope, such that novel fusions such as those described here would be missed.
A multiplex approach to the detection of fusions at the time of diagnosis is recommended for accurate characterization of tumors whose clinical and histologic differential diagnosis includes infantile fibrosarcoma or congenital mesoblastic nephroma. A broader approach will also optimize the identification of patients who could potentially benefit from targeted therapies such as TRK inhibitors. Next-generation sequencing panels that include fusions are increasingly available in academic and commercial laboratories. The targeted RNA sequencing assay used here has the advantage of being able to identify fusion partners from a starting point in only one partner. For example, by sequencing RNA starting at appropriate exon breakpoint regions in NTRK3, one can identify ETV6, EML4, or a novel fusion partner. Furthermore, the assay can be multiplexed, so that samples are screened simultaneously for multiple fusions. For example, a panel could be designed to include both NTRK3 fusions and ETV6 fusions, in addition to NTRK1, NTRK2, and other significant soft tissue tumor fusions. A multiplex sequencing approach is particularly helpful when considering a wide differential diagnosis. A more conservative approach for pathologists who do not have access to sequencing panels would be to conduct an initial ETV6 break-apart FISH study, which is widely available, with a reflex to NTRK3 break-apart FISH if the first result is negative. This approach will require the availability of NTRK3 break-apart FISH.
The primary aim of this analysis was to describe the recurrent nature of the EML4–NTRK3 rearrangement, thus cases with a range of histologic features whose differential diagnosis included infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma or secretory breast carcinoma were included. Cases of infantile fibrosarcoma, in particular, which were diagnosed during the era of routine ETV6–NTRK3 testing did not have a specific diagnosis made if the fusion was not detected, which speaks to the importance of molecular testing in this tumor type. For example, the two cases of infantile fibrosarcoma with EML4–NTRK3 fusions described here were both diagnosed descriptively. Future studies that include comprehensive histopathologic review of large cohorts will be needed to describe the frequency of this rearrangement in each tumor type and any possible differences in behavior and outcome.
Expanding the scope of genetic testing for infantile fibrosarcoma, congenital mesoblastic nephroma, mammary analog secretory carcinoma and secretory breast carcinoma will result in a better understanding of the biology of these tumors and more refined diagnoses for patients. The reliable identification of gene fusion partners is especially important with the emergence of targeted therapy, and will allow for optimal treatment decisions and improved patient outcomes.
References
Chung EB, Enzinger FM . Infantile fibrosarcoma. Cancer 1976;38:729–739.
Blocker S, Koenig J, Ternberg J . Congenital fibrosarcoma. J Pediatr Surg 1987;22:665–670.
Coffin CM, Jaszcz W, O'Shea PA et al. So-called congenital-infantile fibrosarcoma: does it exist and what is it? Pediatr Pathol 1994;14:133–150.
Orbach D, Rey A, Cecchetto G et al. Infantile fibrosarcoma: management based on the european experience. J Clin Oncol 2010;28:318–23.
Soule EH, Pritchard DJ . Fibrosarcoma in infants and children: a review of 110 cases. Cancer 1977;40:1711–1721.
Coulibaly B, Barel E, Soulier M et al. Prenatal diagnosis of infantile fibrosarcoma with diffuse metastases. Prenat Diagn 2008;28:773–775.
Salloum E, Caillaud JM, Flamant F et al. Poor prognosis infantile fibrosarcoma with pathologic features of malignant fibrous histiocytoma after local recurrence. Med Pediatr Oncol 1990;18:295–298.
Grier HE, Perez-Atayde AR, Weinstein HJ . Chemotherapy for inoperable infantile fibrosarcoma. Cancer 1985;56:1507–1510.
Loh ML, Ahn P, Perez-Atayde AR et al. Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children's Hospital, Boston. J Pediatr Hematol Oncol 2002;24:722–726.
Knezevich SR, McFadden DE, Tao W et al. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998;18:184–187.
Gadd S, Beezhold P, Jennings L et al. Mediators of receptor tyrosine kinase activation in infantile fibrosarcoma: a children's oncology group study. J Pathol 2012;228:119–130.
Lannon CL, Martin MJ, Tognon CE et al. A highly conserved NTRK3 C-terminal sequence in the ETV6-NTRK3 oncoprotein binds the phosphotyrosine binding domain of insulin receptor substrate-1: an essential interaction for transformation. J Biol Chem 2004;279:6225–6234.
Sheng WQ, Hisaoka M, Okamoto S et al. Congenital-infantile fibrosarcoma. A clinicopathologic study of 10 cases and molecular detection of the ETV6-NTRK3 fusion transcripts using paraffin-embedded tissues. Am J Clin Pathol 2001;115:348–355.
Bourgeois JM, Knezevich SR, Mathers JA et al. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol 2000;24:937–946.
Dubus P, Coindre JM, Groppi A et al. The detection of tel-TrkC chimeric transcripts is more specific than TrkC immunoreactivity for the diagnosis of congenital fibrosarcoma. J Pathol 2001;193:88–94.
Malkan AD, Loh A, Bahrami A et al. An approach to renal masses in pediatrics. Pediatrics 2015;135:142–158.
Knezevich SR, Garnett MJ, Pysher TJ et al. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res 1998;58:5046–5048.
Rubin BP, Chen CJ, Morgan TW et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol 1998;153:1451–1458.
Tognon C, Knezevich SR, Huntsman D et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002;2:367–376.
Skalova A, Vanecek T, Sima R et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol 2010;34:599–608.
Leeman-Neill RJ, Kelly LM, Liu P et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer 2014;120:799–807.
Seethala RR, Chiosea SI, Liu CZ et al. Clinical and morphologic features of ETV6-NTRK3 translocated papillary thyroid carcinoma in an adult population without radiation exposure. Am J Surg Pathol 2017;41:446–457.
Dogan S, Wang L, Ptashkin RN et al. Mammary analog secretory carcinoma of the thyroid gland: a primary thyroid adenocarcinoma harboring ETV6-NTRK3 fusion. Mod Pathol 2016;29:985–995.
Krings G, Joseph NM, Bean GR et al. Genomic profiling of breast secretory carcinomas reveals distinct genetics from other breast cancers and similarity to mammary analog secretory carcinomas. Mod Pathol 2017;30:1086–1099.
Adem C, Gisselsson D, Dal Cin P et al. ETV6 rearrangements in patients with infantile fibrosarcomas and congenital mesoblastic nephromas by fluorescence in situ hybridization. Mod Pathol 2001;14:1246–1251.
Argani P, Fritsch M, Kadkol SS et al. Detection of the ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: application to challenging pediatric renal stromal tumors. Mod Pathol 2000;13:29–36.
Argani P, Fritsch MK, Shuster AE et al. Reduced sensitivity of paraffin-based RT-PCR assays for ETV6-NTRK3 fusion transcripts in morphologically defined infantile fibrosarcoma. Am J Surg Pathol 2001;25:1461–1464.
Tannenbaum-Dvir S, Glade Bender JL, Church AJ et al. Characterization of a novel fusion gene EML4-NTRK3 in a case of recurrent congenital fibrosarcoma. Cold Spring Harb Mol Case Stud 2015;1:a000471.
Wong V, Pavlick D, Brennan T et al. Evaluation of a congenital infantile fibrosarcoma by comprehensive genomic profiling reveals an LMNA-NTRK1 gene fusion responsive to crizotinib. J Natl Cancer Inst 2015;108:djv307.
Ito Y, Ishibashi K, Masaki A et al. Mammary analogue secretory carcinoma of salivary glands: a clinicopathologic and molecular study including 2 cases harboring ETV6-X fusion. Am J Surg Pathol 2015;39:602–610.
Skalova A, Vanecek T, Simpson RH et al. Mammary analogue secretory carcinoma of salivary glands: molecular analysis of 25 ETV6 gene rearranged tumors with lack of detection of classical ETV6-NTRK3 fusion transcript by standard RT-PCR: report of 4 cases harboring ETV6-X gene fusion. Am J Surg Pathol 2016;40:3–13.
Nagasubramanian R, Wei J, Gordon P et al. Infantile fibrosarcoma with NTRK3-ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr Blood Cancer 2016;63:1468–1470.
Laetsch TW, DuBois SG, Nagasubramanian R et al. A pediatric phase 1 study of larotrectinib, a highly selective inhibitor of the tropomyosin receptor (TRK) family. J Clin Oncol 2017; 10510.
Roberts KG, Bridges O, Janke LJ et al. Genetic modeling and therapeutic targeting of ETV6-NTRK3 with Loxo-101 in acute lymphoblastic leukemia. Blood 2016;128:278.
Shukla N, Roberts S, Baki MO et al. Successful targeted therapy of refractory pediatric ETV6-NTRK3 fusion-positive secretory breast carcinoma. JCO Precision Oncol 2017; available from ascopubs.org/doi/pdf/10.1200/PO.17.00034.
Soda M, Choi YL, Enomoto M et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–566.
Choi YL, Takeuchi K, Soda M et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res 2008;68:4971–4976.
Mitelman F, Johansson B, Mertens F . The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007;7:233–245.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Three authors, Namitha Manoj, Josh D Haimes and Joshua Stahl, are employed by ArcherDX, which supplies the library preparation technology and analysis software used in this study. They receive salary support and are shareholders in the business.
Rights and permissions
About this article

Cite this article
Church, A., Calicchio, M., Nardi, V. et al. Recurrent EML4–NTRK3 fusions in infantile fibrosarcoma and congenital mesoblastic nephroma suggest a revised testing strategy. Mod Pathol 31, 463–473 (2018). https://doi.org/10.1038/modpathol.2017.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.127
This article is cited by
-
Detection of sarcoma fusions by a next-generation sequencing based–ligation-dependent multiplex RT-PCR assay
Modern Pathology (2022)
-
Treatment Patterns of Real-World Patients with TRK Fusion Cancer Treated by US Community Oncologists
Targeted Oncology (2022)
-
Applicability of pan-TRK immunohistochemistry for identification of NTRK fusions in lung carcinoma
Scientific Reports (2021)
-
Novel BRAF gene fusions and activating point mutations in spindle cell sarcomas with histologic overlap with infantile fibrosarcoma
Modern Pathology (2021)
-
Broadening the spectrum of NTRK rearranged mesenchymal tumors and usefulness of pan-TRK immunohistochemistry for identification of NTRK fusions
Modern Pathology (2021)
