
Abstract
The TP63 gene encodes a member of the p53 family of transcription factors. Although TP53 is a well-known tumor suppressor gene, the role of p63 in tumorigenesis is controversial. Our group recently identified novel chromosomal rearrangements involving TP63 in approximately 6% of peripheral T-cell lymphomas, which correlated with a p63+/p40− immunohistochemical profile. As a subset of lung adenocarcinomas are p63+/p40−, we undertook the current study to examine the presence of TP63 rearrangements and correlate with p63/p40 expression. Next-generation sequencing was used to identify genomic rearrangements of TP63 in 37 adenocarcinomas. Confirmatory fluorescence in-situ hybridization (FISH) using a break-apart probe to the TP63 gene region and immunohistochemistry for p63 and p40 were performed on adenocarcinomas with TP63 rearrangements identified by mate-pair sequencing. Immunohistochemistry for p63 and p40 was performed on 45 additional adenocarcinomas, and FISH was performed on all adenocarcinomas with p63 positivity. TP63 rearrangement was identified in two adenocarcinoma specimens from a single patient. The rearrangement resulted in a complex rearrangement of 3q that fused B3GALNT1 at the 3′ intron to TP63. FISH confirmed the rearrangement in both tumors. Immunohistochemistry staining for p63 was diffuse (>80% cells+) and p40 was negative. Of the 44 additional adenocarcinomas, 13 (30%) showed p63 expression; p40 was negative in all cases. No case showed rearrangement of TP63 by a break-apart FISH. However, extra copies of the intact TP63 locus were seen in the p63-positive areas of all 12 cases, with copy numbers ranging from three to seven. We have identified a novel chromosomal rearrangement involving TP63 in a p63+/p40− lung adenocarcinoma. Break-apart FISH testing can be used to diagnose this finding. Immunohistochemistry for p63 was not specific for this rearrangement, as nearly 33% of adenocarcinomas expressed p63. Additional copies of the intact TP63 locus were also a common finding and correlated with immunohistochemistry positivity for p63.
Similar content being viewed by others

Main
The TP63 gene, located on chromosome 3q28, encodes a member of the p53 family of transcription factors.1 The p63 protein has a critical role in the maintenance of progenitor cells in epithelial development and morphogenesis. Indeed, Trp63 knockout mice show severe developmental defects, such as lack of limbs and tissues such as teeth and mammary glands.2 Furthermore, these mice do not develop complete stratified epithelium. Human TP63 variants most commonly encompass single-nucleotide point mutations and have been reported in various genetic syndromes, including Acro-dermal-ungual-lacrimal-tooth syndrome, ectrodactyly-ectodermal dysplasia and cleft lip/palate syndrome, Hay–Wells syndrome, and limb-mammary syndrome.2 Interestingly, none of these syndromes are tumor prone.
Although TP53 is a well-known tumor suppressor gene and its role in tumorigenesis has been extensively studied, the role of p63 in tumorigenesis has been more controversial. Based on in vitro and in vivo mouse studies, full-length p63 containing the N-terminal transactivation domain (TAp63) shares the most homology with p53 and acts as a tumor suppressor gene, inducing growth suppression and death.2, 3, 4 In contrast, p63 isoforms with N-terminal truncations (ΔNp63) may have either tumor suppressor or oncogenic function, depending on the genes that are transactivated.5, 6 Several mouse models with Trp63 mutations are tumor prone. The tumors are of various types, including carcinomas and sarcomas, and usually are metastatic, indicating aggressive behavior.6
TP53 is commonly mutated in human cancers; in contrast, mutations in TP63 have rarely been reported, including in lung cancer (<4% of non-small-cell carcinomas studied).3, 5, 7, 8 Most are missense mutations reportedly with a tumor suppressor activity.3, 8 These mutations have been described in both squamous cell carcinoma and adenocarcinoma.
However, TP63 amplification and immunohistochemical nuclear expression is common in many cancers.5, 9 In lung cancer, amplification and expression is most commonly observed in squamous cell carcinoma, and p63 by immunohistochemistry has been routinely used to distinguish squamous cell carcinoma from adenocarcinoma.10, 11, 12 However, amplification has been reported in a small subset (11%) of adenocarcinoma and expression consistently observed in up to 32% of adenocarcinoma.13, 14, 15 Usually p63 expression in adenocarcinoma is focal but in about 5% of tumors, expression is more diffuse.16, 17 Most antibodies raised against p63, including the 4A4 clone commonly used for immunohistochemistry, do not distinguish between the transactivated and ΔN isoforms of p63; the proposed explanation for the detection of p63 expression in adenocarcinoma. In contrast, an antibody against p40, which targets the ΔN isoforms of p63, has been shown to be highly specific for squamous cell carcinoma and rarely reported in adenocarcinoma.15, 17
Using next-generation sequencing analysis of mate-pair DNA libraries, our group recently identified novel chromosomal rearrangements involving TP63 in approximately 6% in peripheral T-cell lymphomas and demonstrated that these were associated with a poorer overall survival than lymphomas without these rearrangements.18 These rearrangements also were found in occasional B-cell lymphomas, but were not identified by RNA sequencing in a small series of carcinomas of the lung, breast, or head and neck.18, 19 By immunohistochemistry, TP63 rearrangements were associated with a p63+/p40− profile in lymphomas.18 As a subset of lung adenocarcinomas has a p63+/p40− immunoprofile, we examined the presence of TP63 rearrangements and associated p63/p40 expression of lung adenocarcinomas.
Materials and methods
Case Selection
Thirty seven lung adenocarcinomas were sequenced using the mate-pair break-point sequencing protocol (see below). For this study, data were mined for TP63 genomic rearrangements. Fluorescence in-situ hybridization (FISH) and immunohistochemistry were used to further evaluate potential TP63 rearrangements. A validation study was performed on a separate cohort of 45 resected adenocarcinoma including immunohistochemistry for p63 and p40. All cases positive for p63 and negative for p40 and two cases negative for both were selected for further FISH studies. The diagnosis of adenocarcinoma was confirmed using the WHO classification20 and classified according to the new proposed classification of adenocarcinoma.21 The Mayo Clinic Institutional Review Board approved the study.
DNA Isolation and Mate-pair Sequencing
Frozen lung adenocarcinoma sections were cut into 10-micron size and pure cell populations were isolated using the Arcturus PixCell II microscope and CapSure Macro Laser Capture Microdissection caps (Arcturus; LCM 0211). Associated histologically non-neoplastic tissue was also collected by laser capture microdissection from adjacent benign tissue blocks associated with each case. Whole-genome amplification (WGA) was performed directly on laser captured microdissected cells using a single-step procedure.22 Briefly, laser captured microdissected cells were incubated for 10 min in 0.5X Repli-g D2 buffer (6.5 μl) (Qiagen, CA, USA) and then in Repli-g stop solution (3.5 μl). Repli-g mini kit Master Mix (40 μl) was then added and incubated at 30 °C for 16 h. Four individual 50 μl WGA reactions were pooled for each sample. DNA was quantified by Quant-iT PicoGreen analysis (Invitrogen, P7581). Mate-Pair libraries were assembled from WGA DNA according to a previously published protocol22 using the Illumina mate-pair kit. Briefly, WGA DNA (10 μg) was fragmented to ∼3 kb and DNA intramolecular-circles assembled by ligation following biotin-end labeling. Additional fragmentation to 350–650 bp was followed by immobilization of biotinylated terminal fragments on M-280 streptavidin beads (Dynal) and assembly of adapter flanked Illumina-indexed paired-end libraries using Illumina adapters (Illumina, CA, USA). Two multiplexed libraries were loaded per lane of an Illumina flow cell and sequenced to 101 × 2 paired-end reads on an Illumina HiSeq. Base calling was performed using Illumina Pipeline v1.5.
Sequencing Alignment and Bioinformatic Analysis
Bioinformatic protocols to rapidly and efficiently process Next-Generation Sequencing Mate-Pair data using a 32-bit binary indexing of the hg19 reference genome, to which consecutive 32-bit binary sequences from associated Mate Pair reads are aligned, have been previously published from our laboratory.18, 23 The algorithm maps both Mate-Pair reads successively to the whole genome, selecting reads <15 kb apart allowing up to 10 mismatches, with the lowest cumulative mismatch count sent to the output. Discordant Mate-Pairs mapping >15 kb apart or to different chromosomes were selected for further analysis and associated fragments were clustered together. False positives were eliminated using algorithmic filters based on homology scores calculated during mapping. Replication was calculated for each chromosome and replicate read-pairs were removed. Bridged coverage was calculated as the sum of the fragment lengths (distance between read 1 and read 2) of correctly mapping Mate-Pair and Pair-End reads, divided by the mappable chromosome size. Base coverage was calculated from the total number of mappable reads, multiplied by the read length and divided by the mappable chromosome size.
PCR Validation
Primers were designed flanking the predicted TP63 break point (5′-GAATGAAGGA ATGAAATACC CTCAGATGAC-3′; chromosome 3, 160,819,243–160,819,272 and 5′-CTATGAAAAA GCTGGTGCTC ATCAGACG-3′; chromosome 3, 189,430,262–189,430,289). Validation PCR (25 μl reaction volumes, 50 ng Template, 35 cycles) was performed using the EasyA high fidelity polymerase (Stratagene, La Jolla, CA, USA, #600404) on tumor DNA from the adenocarcinomas (AD1 and AD2), non-neoplastic lung from the same patient and a human Genomic DNA control (G304A; Promega, Madison, WI, USA), as well as germline blood DNA (B). PCR products were visualized on 1% agarose gel and excised/extracted DNA was sequenced by standard Sanger sequencing protocols using both forward and reverse primers.
Immunohistochemistry for p63 and p40
Four micron sections cut from formalin-fixed, paraffin-embedded blocks were placed on ‘charged’ slides; slides were then dried and melted in a 62 °C oven for 20 min. Slides were stained with mouse monoclonal p63 antibody (clone BC4A4, Biocare, Concord, CA, USA) at a 1/250 titer, or with rabbit polyclonal p40 antibody (Biocare) at a 1/100 titer. Slides were placed on a Ventana BenchMark XT (Ventana Medical Systems, Oro Valley, AZ, USA) for staining. The staining protocol included online deparaffinization, Heat Induced Epitope Retrieval with Ventana Cell Conditioning 1 for 32 mins, primary antibody incubation for 16 mins (p63) or 32 mins (p40) at 37 °C. Antigen–antibody reactions were visualized using the Ventana OptiViewTM Universal DAB Detection Kit. Counterstaining was performed online using Ventana Hematoxylin II for 8 mins, followed by bluing reagent for 4 mins. All slides were removed from the stainer, dehydrated, and coverslipped for microscopic examination. Appropriate positive and negative controls were used.
Results were reported as percent of tumor cells with nuclear staining, and only cases with >5% positive cells were considered positive. If present, cytoplasmic staining for p63 was noted.
FISH
Interphase FISH was performed on formalin-fixed paraffin-embedded sections of lung adenocarcinoma and controls using a break-apart probe to the TP63 gene region as previously described.18 Briefly, sections were deparaffinized and dehydrated, pretreated with Tris/EDTA and NaCl protease, and hybridized with probes flanking the TP63 gene and fluorescently labeled with Spectrum Orange (5′) or Spectrum Green (3′). Slides were analyzed for abnormal separation of red and green fluorescent signals using standard fluorescence microscopy techniques. In cases with a heterogeneous staining pattern for p63 by immunohistochemistry, p63-positive and p63-negative areas of the tumor were circled and scored separately in the FISH analysis.
Results
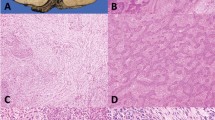
Of the 37 sequenced adenocarcinomas, a TP63 rearrangement was identified in two (5%) synchronous lung tumors from one patient (Figure 1). This patient underwent resection of two adenocarcinomas with a 5 months interval, the first from the right middle lobe and the second from the left lower lobe. Histologically, both tumors were mucinous adenocarcinoma and determined to be intrapulmonary metastasis through shared genomic break points. The TP63 fusion resulted from a complex rearrangement on the q-arm of chromosome 3 that fused the B3GALNT1 gene at locus 3q26.1a to TP63 at locus 3q28b (Figure 1a). The rearrangement predicted an inversion of the gene sequence in the third intron of B3GALNT1 transcript variant 1, which fused with a region in the first intron of TP63 transcript variant 1, positioning the promoter of B3GALNT1 in line with the TP63 coding unit from exon 2 onwards (Figure 1b). PCR validation of the rearrangement using primers flanking the predicted fusion junction generated unique bands in both adenocarcinomas, which was absent in non-neoplastic lung tissue from the same patient and a mixed germline population control sample (Figure 1c). Additional validation in germline DNA extracted from blood from that patient also confirmed the event as a novel somatic rearrangement (Figure 1d). Sanger sequencing on DNA extracted from the PCR validation bands confirmed the fusion with precise positions at chromosome 3, 160,819,605 and 189,429,980, with an intermediate 11 nucleotides between these fusion positions of unknown origin (Figure 1e). Immunohistochemistry staining for p63 was diffuse (>80% tumor cells+) and p40 was negative in both adenocarcinomas (Figure 2a–c). Break-apart FISH confirmed rearrangement of TP63 in both samples (Figure 2d).

(a) Junction plot of Mate-Pair data showing inversion chr3:(q26–q28) involving B3GALNT1 and TP63. Regions 3q26.1 and 3q28b are presented on the upper and lower parts of the plot, respectively. Mate-pair reads are mapped to the genome positions as red or blue dots, according to their mapping polarities to the positive or negative DNA strands, respectively. Normal concordantly mapping mate-pair reads are presented as red/blue linked arrays moving vertically from each region. Discordantly mapping mate-pair reads are presented as dots with linked lines spanning across the upper and lower represented genomic areas. (b) Schematic representation of the predicted B3GALNT1/TP63 fusion. (c) PCR validation on adenocarcinomas (AD1 and AD2), non-neoplastic lung (NL), and mixed population germline control (Con). (d) Additional PCR validation on blood-derived germline DNA (B). (e) Sanger sequencing profile showing fusion junction.

(a) Intermediate power magnification of one of the mucinous adenocarcinomas with TP63 rearrangement (H&E, × 200). (b) The adenocarcinoma is diffusely positive for p63 (p63, × 200), (c) and negative for p40 (p40, × 200). (d) The break-apart FISH for p63 show an abnormal separation of the orange and green fluorescent signals indicative of the chromosomal rearrangement.
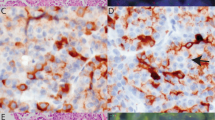
Of the 45 additional adenocarcinomas, 13 of 44 (one technical failure, 30%) showed p63 expression by immunohistochemistry in 20–60% of cells; p40 was negative in all cases (Table 1). None of the cases showed cytoplasmic staining. FISH was performed in 12 of these 13 cases (one had insufficient tissue). No case showed rearrangement of TP63 by break-apart FISH. However, extra copies of the intact TP63 locus were seen in the p63-positive areas of all 12 cases, with copy numbers ranging from 3 to 7 (Figure 3). In four cases, the areas of immunohistochemical positivity for p63 were sufficiently discrete that FISH could be scored separately in the p63-positive and p63-negative areas. Although all p63-positive areas demonstrated extra copies of TP63, only two copies of TP63 were present in the p63-negative areas in three of four cases. FISH was also performed in two adenocarcinomas negative for both p63 and p40, and only two copies of TP63 were found.

(a) Adenocarcinoma with focal expression of p63 (p63, × 200). (b) FISH demonstrates extra copies of the intact TP63 locus in this area. (c and d) In contrast, in the area of adenocarcinoma with no expression of p63, only two copies of TP63 are identified by FISH.
The TP63 rearrangement was identified in a multifocal mucinous adenocarcinoma. Further studies of two additional mucinous adenocarcinomas failed to show p63 expression. P63 expression was less common in lepidic predominant adenocarcinoma than other adenocarcinoma subtypes (Table 1).
Discussion
To our knowledge, this is the first report of chromosomal rearrangements of TP63 in lung cancer, specifically in lung adenocarcinoma. Using the same strategy as in peripheral T-cell lymphoma,18 we first identified a 3q26–28 chromosomal rearrangement in sequenced lung adenocarcinoma, which validated using PCR and Sanger sequencing. We further confirmed the rearrangements using a break-apart FISH probe, and also performed immunohistochemistry for p63 and p40. Thus far, TP63 genomic rearrangements had been described only in lymphomas.18, 19 In diffuse large B-cell lymphomas, TP63 is fused to TBL1XR1. This fusion is seen in 5% of diffuse large B-cell lymphomas, exclusively of germinal center type.19 The authors hypothesized that the fusion was functionally similar to the ΔNp63, potentially providing a proliferative advantage and resistance to genotoxic stress induced by chemotherapy. Vasmatzis et al18 reported TP63 rearrangements in 11 (5.8%) peripheral T-cell lymphomas, seven of which had TBL1XR1 as a partner, and in two (1.2%) diffuse large B-cell lymphomas. The authors hypothesized that TP63 rearrangements may represent an alternate route of deregulating the p53 pathway in peripheral T-cell lymphoma. Interestingly, the authors did not detect any TP63 fusions using the Snow-Shoes-FTD algorithm in transcriptome sequencing data in the studied lung cancers. However, the histologic subtypes of these lung cancers were unknown.
In our patient’s tumors, the partner to TP63 is B3GALNT1. This gene is a member of the β-1,3-galactosyltransferase gene family which encodes for membrane-bound glycoproteins with various enzymatic functions. According to the catalog of somatic mutations in cancer, mutations in B3GALNT1 are uncommon (0.4%) and mostly missense. In the Cancer Genome Atlas database, four mutations (three noncoding and one missense) were identified in adenocarcinoma and one (noncoding) in squamous cell carcinoma. No fusion transcript involving this gene has been reported. The role of TP63 rearrangement in lung carcinogenesis still remains to be elucidated. The findings of TP63 amplification and p63 expression in lung cancer suggest a role for TP63 in cancer development. Indeed, TP63 amplification is present in high-grade dysplasia/squamous cell carcinoma in situ but not in low-grade dysplasia.14 Similarly, p63 expression progressively increases from pre-neoplastic to pre-invasive and invasive squamous cell carcinoma.13
The clinical significance of a chromosomal rearrangement of TP63 in lung adenocarcinoma will need to be studied in a large cohort of patients as the frequency of this finding appears low, seen in only one (1%) patient from our cohort of 81 patients, although this is a frequency similar to other clinically relevant genomic events in lung cancer. In lymphomas, TP63 rearrangements are associated with adverse outcomes. In diffuse large B-cell lymphoma, three of five TP63 rearranged cases were refractory to R-CHOP chemotherapy.19 In peripheral T-cell lymphoma, TP63 rearranged cases had a significantly worse overall survival.18 In lung cancer, the role of TP63 amplification and p63 expression on outcome is not clear. Massion et al14 suggested that TP63 amplification conferred a survival advantage to patients with non-small-cell carcinoma when compared with the overall population as well as patients with squamous cell carcinoma. Similarly, they found a strong association between the intensity of p63 expression and the odds of surviving, even after adjusting for various prognostic parameters such as stage. Furthermore, Au et al24 showed that squamous cell carcinomas expressing p63 had a better outcome when compared with p63-negative squamous cell carcinomas; however, this prognostic significance was lost when non-small-cell carcinomas were analyzed as a group. In other studies, p63 and p40 expressions did not appear to affect overall survival or disease-free survival in non-small-cell carcinoma.13, 25
Given that p40 is negative in lung adenocarcinomas, the expression of p63 has been attributed to the presence of the transactivated isoform.15 However, this is not necessarily the case. The TP63 rearrangements we previously described in peripheral T-cell lymphomas encode fusion proteins homologous to ΔNp63, in which they lack the transactivation domain of p63.18 In addition to lacking this transactivation domain, the native transcripts that encode ΔNp63 isoforms also differ from TAp63 transcripts in which they are regulated by a distinct promoter region and encode a short-leader sequence (so-called ‘TA*’) N-terminal to the DNA-binding domain of the resultant protein. The anti-p40 antibody recognizes this short-leader sequence. In contrast, p63 fusion proteins lack this leader sequence. In peripheral T-cell lymphomas, these proteins contain portions of the fusion partner fused directly to the DNA-binding domain of p63 (encoded by exons 4–8), and the resultant fusion protein is non-reactive with the anti-p40 antibody in lymphoma (ALF, unpublished). In the current lung cancer case, the DNA sequence data predict a fusion protein encoded by exons 1–3 of B3GALNT1 fused to a truncated TAp63 protein lacking the N-terminal portion of the transactivation domain (encoded by exon 1 of TP63). In contrast, the 4A4 antibody commonly used in immunohistochemistry is derived against an epitope spanning regions of the p63 protein common to all native isoforms as well as the described fusion proteins. Thus, this antibody cannot distinguish native ΔNp63 from TAp63 isoforms, nor native protein from fusion protein. Therefore, TP63 rearrangements possibly could explain expression of p63 in a subset of adenocarcinoma, particularly cases with diffuse expression as seen in our case.
p63 expression in lung adenocarcinoma could also be explained by amplification of TP63. In our study, all adenocarcinomas expressing p63 that did not have rearrangements of TP63 demonstrated increased copy numbers of TP63. Interestingly, this correlated with the focal expression of p63. Indeed, in cases where FISH could be scored separately in the p63-positive and p63-negative areas, increased copy numbers were observed only in the p63-positive areas. Additionally, no increased copy numbers was identified in the two p63/p40-negative adenocarcinomas. The correlation between p63 expression and TP63 amplification is well demonstrated in lung squamous cell carcinoma and has also been reported in lung adenocarcinoma, although with lower frequency.5, 14 In our study, a third of the adenocarcinoma demonstrated p63 expression and increased copy numbers of TP63. This is in contrast to the 0–11% reported frequency of TP63 amplification.5, 14 Reported expression of p63 in adenocarcinoma has been more variable but similar to our results, ranging up to 31%,10, 11, 12, 13, 14, 15, 16, 17 with most recent studies focusing only on expression and not amplification, which could explain the lower reported amplification frequency.
References
Senoo M, Seki N, Ohira M, et al. A second p53-related protein, p73l, with high homology to p73. Biochem Biophys Res Commun 1998;248:603–607.
Brunner HG, Hamel BCJ, van Bokhoven H The p63 gene in EEC and other syndromes. J Med Genet 2001;39:377–381.
Osada M, Ohba M, Kawahara C, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med 1998;4:839–843.
Kato S, Shimada A, Osada M, et al. Effects of p51/p63 missense mutations on transcriptional activities of p53 downstream gene promoters. Cancer Res 1999;59:5908–5911.
Hibi K, Trink B, Patturajan M, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA 2000;97:5462–5467.
Su X, Chakravarti D, Flores ER p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Med 2013;13:136–142.
Hagiwara K, McMenamin G, Miura K, et al. Mutational analysis of the p63/p73Lp51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Res 1999;59:4165–4169.
Sunahara M, Shishikura T, Takahashi M, et al. Mutational analysis of p51A/Tap63γ, a p53 homolog, in non-small cell lung cancer and breast cancer. Oncogene 1999;18:3761–3765.
Nylander K, Vojtesek B, Nenutil R, et al. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J Pathol 2002;198:417–427.
Kaufmann O, Fietze E, Mengs J, et al. Value of p63 and CK5/6 as immunohistochemical markers for the differential diagnosis of poorly differentiated and undifferentiated carcinomas. Am J Clin Pathol 2001;116:823–830.
Kargi A, Gurel D, Tuna B The diagnostic value of TTF-1, CK5/6, and p63 immunostaining in the classification of lung carcinoma. Appl Immunohistochem Mol Morphol 2007;15:415–420.
Mukhopadhyay S, Katzenstein A-L Subclassification of non-small cell lung carcinomas lacking morphologic differentiation on biopsy specimens: utility of an immunohistochemical panel containing TTF-1, Napsin A, p63, and CK5/6. Am J Surg Pathol 2011;35:15–25.
Pelosi G, Pasini F, Olsen Stenholm C, et al. p63 immunoreactivity in lung cancer: yet another player in the development of squamous cell carcinomas?. J Pathol 2002;198:100–109.
Massion PP, Taflan PM, Jamshedur Rahman SM, et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res 2003;63:7113–7121.
Nonaka D A study of ΔNp63 expression in lung non-small cell carcinomas. Am J Surg Pathol 2012;36:895–899.
Rekhtman N, Ang DC, Sima CS, et al. Immunohistochemical algorithm for differentiation of lung adenocarcinoma and squamous cell carcinoma based on large series of whole-tissue sections with validation in small specimens. Mod Pathol 2011;24:1348–1359.
Bishop JA, Teruya-Feldstein J, Westra WH, et al. p40 (ΔNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma. Mod Pathol 2012;25:405–415.
Vasmatzis G, Johnson SH, Knudson RA, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood 2012;120:2280–2289.
Scott DW, Mungall KL, Ben-Neriah S, et al. TBL1XR1/TP63: a novel recurrent gene fusion in B-cell non-Hodgkin lymphoma. Blood 2012;119:4949–4952.
Travis WD, Brambilla E, Muller-Hermelink KH, Harris CC eds. Tumors of the Lung, Pleura, Thymus and Heart. WHO classification of tumors. IARC Press: Lyon. 2004;37–46.
Travis WD, Brambilla E, Noguchi M, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma. J Thorac Oncol 2011;6:244–285.
Murphy SJ, Cheville JC, Zarei S, et al. Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA Res 2012;19:395–406.
Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6,7)(p25 3,q32 3) translocations in ALK negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011;117:915–919.
Au NHC, Cheang M, Hunstman DG, et al. Evaluation of immunohistochemical markers in non-small cell lung cancer by unsupervised hierarchical clustering analysis: a tissue microarray study of 284 cases and 18 markers. J Pathol 2004;204:101–109.
Iwata T, Uramoto H, Sugio K, et al. A lack of prognostic significance regarding ΔNp63 immunoreactivity in lung cancer. Lung Cancer 2005;50:67–73.
Acknowledgements
This work was supported by the Mayo Clinic Center for Individualized Medicine (CIM) Biomarker Discovery (BMD) Program. ALF is a Damon Runyon Clinical Investigator supported by the Damon Runyon Cancer Research Foundation (CI-48-09).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Aubry, MC., Roden, A., Murphy, S. et al. Chromosomal rearrangements and copy number abnormalities of TP63 correlate with p63 protein expression in lung adenocarcinoma. Mod Pathol 28, 359–366 (2015). https://doi.org/10.1038/modpathol.2014.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.118
This article is cited by
-
p63 expression in human tumors and normal tissues: a tissue microarray study on 10,200 tumors
Biomarker Research (2021)
-
p63 isoforms in triple-negative breast cancer: ΔNp63 associates with the basal phenotype whereas TAp63 associates with androgen receptor, lack of BRCA mutation, PTEN and improved survival
Virchows Archiv (2018)
