
Abstract
Merkel cell carcinoma is a highly aggressive cutaneous neuroendocrine tumor that has been associated with Merkel cell polyomavirus in up to 80% of cases. Merkel cell polyomavirus is believed to influence pathogenesis, at least in part, through expression of the large T antigen, which includes a retinoblastoma protein-binding domain. However, there appears to be significant clinical and morphological overlap between polyomavirus-positive and polyomavirus-negative Merkel cell carcinoma cases. Although much of the recent focus of Merkel cell carcinoma pathogenesis has been on polyomavirus, the pathogenesis of polyomavirus-negative cases is still poorly understood. We hypothesized that there are underlying human somatic mutations that unify Merkel cell carcinoma pathogenesis across polyomavirus status, and to investigate we performed whole exome sequencing on five polyomavirus-positive cases and three polyomavirus-negative cases. We found that there were no significant differences in the overall number of single-nucleotide variations, copy number variations, insertion/deletions, and chromosomal rearrangements when comparing polyomavirus-positive to polyomavirus-negative cases. However, we did find that the retinoblastoma pathway genes harbored a high number of mutations in Merkel cell carcinoma. Furthermore, the retinoblastoma gene (RB1) was found to have nonsense truncating protein mutations in all three polyomavirus-negative cases; no such mutations were found in the polyomavirus-positive cases. In all eight cases, the retinoblastoma pathway dysregulation was confirmed by immunohistochemistry. Although polyomavirus-positive Merkel cell carcinoma is believed to undergo retinoblastoma dysregulation through viral large T antigen expression, our findings demonstrate that somatic mutations in polyomavirus-negative Merkel cell carcinoma lead to retinoblastoma dysregulation through an alternative pathway. This novel finding suggests that the retinoblastoma pathway dysregulation leads to an overlapping Merkel cell carcinoma phenotype and that oncogenesis occurs through either a polyomavirus-dependent (viral large T antigen expression) or polyomavirus-independent (host somatic mutation) mechanism.
Similar content being viewed by others


Main
Merkel cell carcinoma is a rare neuroendocrine tumor of the skin with an aggressive clinical course and an increased prevalence in the elderly and immunosuppressed.1 The incidence of Merkel cell carcinoma has increased in the last several decades, and the United States has an estimated incidence rate of 0.32 per 100 000 persons per year.2 Merkel cell carcinoma has a predilection for sun exposed areas, most often occurring in the head and neck region.3 There is an overall 5-year survival rate of 40%, with stage being a significant prognosticator.3, 4 Merkel cell polyomavirus was discovered in Merkel cell carcinoma and found to be clonally integrated in ∼80% of cases.5, 6, 7 Merkel cell polyomavirus has a high seroprevalence in the general population and asymptomatic infection begins in childhood.8, 9, 10, 11 As one of the steps in the proposed mechanism for Merkel cell carcinoma oncogenesis, polyomavirus must integrate into the human genome.12 Viral integration sites occur throughout the human genome without apparent specificity.13, 14 Emerging data implicate maintenance and expression of the polyomavirus large T antigen in cell cycle dysregulation and the pathogenesis of viral transformation leading to Merkel cell carcinoma.12, 15, 16, 17, 18, 19, 20, 21 Although the molecular role of Merkel cell polyomavirus is quickly evolving, the overall biology of Merkel cell carcinoma is poorly understood. Moreover, the presence of polyomavirus alone is not sufficient for carcinogenesis, namely in those Merkel cell carcinoma cases considered as polyomavirus-negative.
With established subpopulations of polyomavirus-positive and polyomavirus-negative Merkel cell carcinoma, there has been interest in defining what similarities and differences exist between these groups, especially with regard to clinical outcomes and histology. To date, there is somewhat controversial data regarding the clinical outcomes between polyomavirus status Merkel cell carcinoma subpopulations. An early Finnish study of 114 Merkel cell carcinoma samples by Sihtu et al7 showed that polyomavirus-positive cases had a significantly higher (threefold) 5-year overall survival when compared with polyomavirus-negative cases. In a smaller United States study (23 cases) by Bhatia et al,22 median survival was approximately fourfold longer in polyomavirus-positive cases (86 months) than in polyomavirus-negative cases (20 months). In contrast, three subsequent studies by Handschel et al23 (44 German cases), Schrama et al24 (146 Australian and German cases), and Asioli et al25 (70 Italian cases) demonstrated that 5-year overall survival is independent of Merkel cell polyomavirus status. Additionally, it does not appear that polyomavirus status influences recurrence-free survival.24 Although there have been reported subtle, yet statistically significant, nuclear and cytoplasmic differences between polyomavirus-positive and polyomavirus-negative Merkel cell carcinoma as detected by complex morphologic analysis,26 the two are fairly indistinguishable in routine pathological examination of Merkel cell carcinoma by light microscopy.
Despite the conflicting data regarding the outcome and Merkel cell polyomavirus status, it is clear that there is a phenotypic, morphologic, and clinical overlap between polyomavirus-positive and polyomavirus-negative cases. Although Merkel cell polyomavirus is believed to have a role in the majority of cases, a significant proportion (∼20%) of Merkel cell carcinoma is polyomavirus-negative. The existence of Merkel cell polyomavirus-negative cases demonstrates that polyomavirus alone is not sufficient for the development of Merkel cell carcinoma. Thus, it is likely that the Merkel cell carcinoma phenotype develops through distinct, yet convergent, polyomavirus-dependent and polyomavirus-independent mechanisms. The molecular and cellular determinants of these convergent phenotypes have yet to be fully established. In this study, we tested the hypothesis that there are somatically acquired mutations in the human genome, which lead to overlapping morphological and clinical Merkel cell carcinoma phenotypes. We performed whole exome sequencing of polyomavirus-positive and polyomavirus-negative cases as an unbiased approach to detect recurrent somatic mutations in Merkel cell carcinoma and to investigate the possible role of known cancer pathways in Merkel cell carcinoma development.
Materials and methods
Case Selection
The use of human subject material was performed in accordance with guidelines set by the Institutional Review Board of Washington University. Eight total cases from deceased patients were selected for whole exome sequencing from previously published and characterized Merkel cell carcinoma cases for which sufficient tissue from formalin-fixed paraffin-embedded blocks was available for DNA testing and confirmatory studies.13, 27 Clinical characteristics included five patients with metastatic Merkel cell carcinoma and three patients with no reported metastases (summarized in Table 1).
Merkel Cell Polyomavirus Detection
Total genomic DNA was extracted from formalin-fixed paraffin-embedded tissue blocks as previously described.13 To determine Merkel cell polyomavirus status, standard polymerase chain reaction was performed using previously published protocols. Briefly, thermocycler conditions were as follows: (1) 55 °C, 1 min; (2) 95 °C, 1 min; (3) 95 °C, 15 sec; (4) 55 °C, 1 min; and (5) repeat steps 3 and 4 for 35 cycles. Platinum Taq HF (Invitrogen, Grand Island, NY, USA) was used. The MCVPS1 primer set was used to detect Merkel cell polyomavirus as previously published;5 forward sequence 5′-TCAGCGTCCCAGGCTTCAGA-3′, reverse sequence 5′-TGGTGGTCTCCTCTCTGCTACTG-3′. A similarly sized beta-globin product (110 bp) was used as an amplification control (forward sequence 5′-ACACAACTGTGTTCACTAGC-3′; reverse sequence 5′-CAACTTCATCCACGTTCACC-3′). Cloned viral plasmid DNA (pMCV-R17a) was used as a positive control, and no template reactions and DNA from normal controls were used as negative controls. Polymerase chain reaction products were detected by agarose gel electrophoresis following ethidium bromide staining. Five of the cases had reliably detected Merkel cell polyomavirus by standard polymerase chain reaction (06, 18, 24, 27, and 39) and were considered polyomavirus-positive cases. The other three cases (21, 29, and 33) were polyomavirus-negative.
To ensure maximal viral sensitivity, we further determined the Merkel cell polyomavirus copy number using a sensitive, previously published real-time polymerase chain reaction assay.22, 27, 28 Briefly, thermocycler conditions were as follows: (1) 50 °C, 1 min; (2) 95 °C, 1 min; (3) 95 °C, 15 sec; (4) 60 °C, 1 min; and (5) repeat steps 3 and 4 for 40 cycles. Applied Biosystems (Carlsbad, CA, USA) ABI Taqman assay primers were used for the conserved Merkel cell polyomavirus small T antigen, with the following primer sequences: forward sequence 5′-GCAAAAAAACTGTCTGACGTGG-3′; reverse sequence 5′-CCACCAGTCAAAACTTTCCCA-3′; probe sequence 5′-TATCAGTGCTTTATTCTTTGGTTTGGATTTCCTCCT-3′. Cloned MCPyV (pMCV-R17a) viral plasmid was serially diluted and used as the positive control, and no template reactions were used for the negative control. The detection threshold was determined from the threshold cycle (Ct) of the most diluted positive control. The sensitivity of this assay is estimated to be ∼0.0004 viral copies/cell.27, 28
Whole Exome Sequencing
One microgram of total genomic DNA (as determined by Qubit (Life Technologies, Grand Island, NY, USA)) from each of the eight Merkel cell carcinoma cases was first fragmented to ∼200–300 base pairs using a Covaris E210 instrument (Covaris Inc., Woburn, MA, USA) then end-repaired and ligated to universal Illumina sequencing adapters. Sequencing libraries were then hybridized to Agilent V4 exome capture probes as per the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA, USA). Captured DNA was then subjected to limited cycle polymerase chain reaction amplification (eight cycles) using primers with seven base pair sequence indexes to permit multiplex sequencing. DNA from two to three cases was then pooled in equimolar volumes and each pool was sequenced on a HighSeq2000 lane using 2 × 101 base pair paired end reads. Base calls and quality scores were generated by the included Casava software (v1.8).
Data Analysis
The resulting FASTQ files were aligned to NBCI build 37.2 of the human reference genome (hg19) using Novoalign (Novocraft, Selangor, Malaysia) with default paired-end parameters. Quality metrics were then calculated using a variety of publicly available software and sequence data were ‘cleaned’ to mark duplicate reads, recalibrate quality scores, and realigned around known polymorphisms using the Genome Analysis Toolkit (GATK v1.6)29, 30 (http://www.broadinstitute.org/gatk) and picard tools (http://picard.sourceforge.net). Sequence variation was identified using multiple software tools to capture the full spectrum of DNA variation; single-nucleotide polymorphisms and small (<10 base pairs) insertions and deletions (indels) were determined using samtools31 (http://samtools.sourceforge.net) to ensure a low false-positive rate; larger indels (>10 base pairs) were identified using Pindel32 (https://trac.nbic.nl/pindel); translocations were identified using Breakdancer33 (http://breakdancer.sourceforge.net); and copy number variation was identified using CONTRA34 (http://contra-cnv.sourceforge.net). Sequence variants were then annotated using the SeattleSeq annotation server (http://gvsbatch.gs.washington.edu/SeattleSeqAnnotation137/index.jsp) and only variants representing coding region changes in at least one transcript, and not present as constitutional polymorphisms in dbSNP (v130) (http://www.ncbi.nlm.nih.gov/projects/SNP), were considered for further analysis. The resulting novel coding region changes were compared with previously published somatic cancer mutations using the COSMIC database (v64)35, 36 (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic). Further, to ensure that previously described somatic variants were not inadvertently removed from the analysis by filtering known polymorphisms in dbSNP, all detected single-nucleotide variations were compared with COSMIC and manually reviewed.
As paired normal tissue was not available for analysis, we could not directly differentiate between coding region changes representing ‘personal single-nucleotide polymorphisms’ (rare mean allele frequency variants not present in dbSNP) and true somatic mutations. Therefore, we used mutation recurrence among all eight cases to determine which genes were most likely implicated in Merkel cell carcinoma pathogenesis. Coding region variants not present in dbSNP were compared at the gene level (any mutation in the gene) among all cases and segregated by Merkel cell polyomavirus status using custom R scripts (available upon request). High frequency recurrent single-nucleotide variations (present in >five of eight cases) were further filtered against a laboratory-generated ‘blacklist’ of false-positive variants resulting from sequence alignment errors particular to the Agilent V4 exome capture probes. The observed allele fractions of mutations were used to infer co-occurring mutations as has been previously described.37
Fluorescence In Situ Hybridization
Interphase fluorescence in situ hybridization for RB1 was performed on formalin-fixed paraffin-embedded tissue sections cut at a thickness of 5 μm on positively charged microscope slides. The paraffin was removed from the sections with three washes of 5 min each in CitriSolve. The slides were then hydrated in two washes of absolute ethanol for 1 min each and allowed to air dry. The slides were processed through a pretreatment solution of sodium thiocyanate that had been preheated to 80 °C. After a 3 min wash in distilled water, the tissue was digested in protease solution (pepsin in 0.2N HCl) for 15 min at 37 °C, followed by another 3 min wash in distilled water. The slides were allowed to air dry after which they were dehydrated by passing through consecutive 70, 85, and 100 ethanol solutions for 1 min each. The slides were again allowed to air dry before applying prepared probe mixture. Probes used were purchased from Abbott Molecular (Des Plaines, IL, USA) and included Vysis LSI 13 (RB1) 13q14 SpectrumOrange Probe (Catalog no. 05J15-011) and Vysis 13q34 SpectrumGreen fluorescence in situ hybridization Probe Kit-CE (Catalog no. 05N34-020). Probes were diluted at a concentration of 1:50 in tDenHyb-2 hybridization buffer (Insitus Biotechnologies Inc., Albuquerque, NM, USA) and well-mixed. Next, the probe in buffer was applied to the appropriate slide to cover the tissue section and the section was coverslipped. Co-denaturation was achieved by incubating the slides at 73 °C for 5 min in a slide moat. Hybridization occurred by transferring the slides to a 37 °C slide light-shielded, humid slide moat overnight. Post hybridization, the coverslips were removed and the slides immersed in 75 °C wash solution (2XSSC/0.3%NP40) for 2 min followed by a 1 min wash in jar containing the same solution at room temperature. The slides were allowed to air dry in the dark and were then counterstained with 10 μl of DAPI II (Abbott Molecular Inc.). Slides were examined using an Olympus BX60 fluorescent microscope with appropriate filters for SpectrumOrange, SpectrumGreen, and the DAPI counterstain. The signal patterns were documented using a CoolSnap camera and CytoVision Imaging System.
Immunohistochemistry
Immunohistochemistry utilized formalin-fixed paraffin-embedded tissue cut at 5 μm sections and floated onto charged slides. Immunohistochemistry for the retinoblastoma protein was performed by Clarient Inc. (Aliso Viejo, CA, USA). Primary antibodies used included retinoblastoma antibody (clone G3-245; BD Biosciences San Jose, CA, USA) at 1:300 dilution for 30 min and phospho-retinoblastoma (Ser807/811) antibody (Catalog 9308; Cell Signaling Technology, Boston, MA, USA) at 1:200 dilution for 1 h. Automated staining was performed using the Bond-III Autostainer (Leica, Buffalo Grove, IL, USA) for retinoblastoma and Ventana Benchmark XT (Ventana Medical, Tucson, AZ, USA) for phospho-retinoblastoma according to the manufacturer’s protocol. Pretreatment antigen retrieval strategies included Leica Bond Epitope Retrieval solution 2 (EDTA-based buffer, pH 9.0) for retinoblastoma and a protein citrate buffer (pH 6.0) for phospho-retinoblastoma at 100 °C. Breast cancer specimens were used as positive staining controls.
Statistics
Comparisons made between groups were performed using the GraphPad Prism software (La Jolla, CA, USA) and the R statistics package (R, version 2.15.1, R Project for Statistical Computing, http://www.r-project.org/). P-values were determined by the use of the two-tailed unpaired t-test. Plots were created in R.
Results
Demographics and Sequence Metrics
Eight cases of Merkel cell carcinoma that have been previously reported were included in this study (representative Merkel cell carcinoma histology shown in Figure 1).13, 27 Demographic information is summarized in Table 1. Five cases were Merkel cell polyomavirus-positive and the other three were polyomavirus-negative as determined by using the MCVPS1 primer set and agarose gel detection. The average age at diagnosis between the polyomavirus-positive cases (73 years, ±15 years) and the polyomavirus-negative cases (62 years, ±10 years) was not significantly different (P=0.2143). Similarly, the length of time from diagnosis to death between polyomavirus-positive cases (4 years, ±4 years) and the polyomavirus-negative cases (6 years, ±6 years) was not significantly different (P=0.4821). All eight cases had a primary site located within the skin and five of eight cases showed metastases to regional lymph nodes. All three of the polyomavirus-negative cases had metastatic disease, whereas only two of the five polyomavirus-positive cases had metastases. Although a small number of cases, this is consistent with some of the controversial literature reporting a more aggressive clinical course related to polyomavirus-negative cases.7, 22, 38

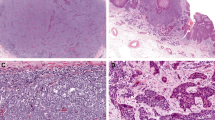
Representative hematoxylin and eosin-stained sections of Merkel cell carcinoma. The tumor comprises sheets and nests of infiltrative high-grade neuroendocrine carcinoma. (a) Low power ( × 100 original magnification). (b) High power ( × 400 original magnification).
Whole human exome sequencing was performed on each of the eight Merkel cell carcinoma cases using formalin-fixed paraffin-embedded tissue. The average number of reads generated per case was 130 477 404 (26 GBases). On average, 94.1% of the exome for each case had at least 25 × coverage. The spectrum of mutations identified include single-nucleotide variations, copy number variations, indels (insertion/deletions), and structural variations. The total number of mutations identified, as well as the type, for each case is summarized in Table 2. Each case generated an average of 41 768 single-nucleotide variation calls, of which 989 represented novel (not in dbSNP) nonsynonymous variants. There was no significant difference in the total number of variants or novel nonsynonymous variants between polyomavirus-positive and polyomavirus-negative groups (P=0.5500 and P=0.0632, respectively). Small indels (as called by samtools) averaged 400 per case and showed no significant difference between polyomavirus-positive and polyomavirus-negative groups (P=0.5350). Larger indels (as called by Pindel) averaged 56 per case and showed no significant difference between polyomavirus-positive and polyomavirus-negative groups (P=0.2424). Similarly, copy number variations (as called by CONTRA) averaged 269 per case and showed no significant difference between polyomavirus-positive and polyomavirus-negative groups (P=0.3919).
Identification of Recurrent Mutation
As paired normal samples were not sequenced along with the Merkel cell carcinoma samples, we could not differentiate between non-pathogenic ‘personal single-nucleotide polymorphisms’ (low mean allele frequency variants specific to an individual and not present in dbSNP) and true somatically acquired variants. Therefore, we looked for recurrent mutations among the eight Merkel cell carcinoma cases to determine genes critical to pathogenesis. Recurrence was assessed at the gene level (ie, the presence or absence of mutations in TP53). Recurrent mutations are summarized in Table 3. There were eight highly recurrent nonsynonymous, non-dbSNP gene variants, present in all eight cases. None of these occurred in known cancer-related genes and the same variant for each gene was observed in each case; all were flagged as ‘blacklisted’ variants representing sequence capture artifact, and these variants were not further analyzed, but are included in the Table 3 for completeness. Analysis of highly recurrent gene variants by Merkel cell polyomavirus status showed that no variants were specific to polyomavirus-positive Merkel cell carcinoma, whereas several genes were associated with polyomavirus-negative Merkel cell carcinoma, including the conical tumor suppressor RB1. All three polyomavirus-negative cases showed truncating, nonsense RB1 mutations including chr13:g.48916767G>A (p.W99*, AF=0.93), (chr13:g.48923137G>A, p.W195*, AF=0.77), and chr13:g.49027222C>T (p.Q597*, AF=0.86) located in exons 3, 6, and 18, respectively. All of these nonsense mutations were predicted to be deleterious by PolyPhen and one (p.W195*) has been previously described in breast cancer (COSMIC ID: COSS1659943).39 To assess the significance of these findings, we compared the rate of truncating, nonsense RB1 mutations previously reported as somatic variants in all cancer types using the COSMIC database (181 of 12 584 cases) to our data. Assuming a rate of truncating mutation in RB1 of 0.014 for both virus-positive and -negative cases of Merkel cell carcinoma, and considering all possible 2 × 2 tables with row sums fixed to 5 and 3 (the numbers of polyomavirus-positive and -negative cases), we obtain a highly significant P-value of 5.6 × 10−6 for the finding of nonsense RB1 mutations in all three polyomavirus-negative cases, but none of the five polyomavirus-positive cases.
Identification of RB1 and RB1 Pathway Mutations
On the basis of the finding of truncating RB1 nonsense mutations in three of three polyomavirus-negative Merkel cell carcinoma cases, we sought to determine whether retinoblastoma-related pathway genes were mutated in polyomavirus-positive cases. Further, although the initial recurrence analysis focused only on single base pair variants, we subsequently analyzed a full range of DNA variation including indels, copy number variations, and translocations, to determine whether retinoblastoma pathway mutations were included in a broader class of mutations. Retinoblastoma pathway mutations are summarized in Table 4. All eight Merkel cell carcinoma cases showed sequence changes predicted to affect at least one of the 14 retinoblastoma pathway genes. The retinoblastoma gene (RB1) itself, was disrupted in five of the eight cases (including the three previously described nonsense mutations, one case with a frame-shift deletion, and one case with RB1 copy loss). The range of RB1 mutations are summarized in Table 5. Strikingly, all three polyomavirus-negative cases had truncating single-nucleotide variant nonsense mutations, which were located in three separate locations of RB1; all mutations were present with a high variant allele fraction. None of the polyomavirus-positive cases had a nonsense RB1 mutation. Two cases, one polyomavirus-positive and one polyomavirus-negative case, each had a large RB1 deletion, and one polyomavirus-positive case had a small deletion involving RB1.
Validation of RB1 pathway mutations
Following the discovery phase of mutations in Merkel cell carcinoma, the DNA and protein-level validation of the changes in RB1 observed in whole exome sequencing was performed. First, the RB1 copy number loss detected in two Merkel cell carcinoma cases by whole exome sequencing was confirmed by fluorescence in situ hybridization (Figure 2). Second, to determine whether mutations detected in the RB1 gene resulted in a corresponding change in retinoblastoma protein expression, we performed immunohistochemistry for the retinoblastoma protein. Retinoblastoma protein showed absent immunoreactivity in each of the five cases with a RB1 mutation discovered by whole exome sequencing (Figure 3); however, the three cases without RB1 mutations by sequencing, showed strong and diffuse nuclear reactivity for the retinoblastoma protein. These results support a one to one relationship with RB1 genetic mutations and corresponding absence of protein. To further explore the retinoblastoma pathway in Merkel cell carcinoma, immunohistochemistry for phosphorylated-retinoblastoma protein was performed. Under normal regulatory circumstances in actively cycling tumor cells, retinoblastoma protein is phosphorylated, leading to its inactivation as a tumor suppressor, S-phase entry, and cell division. Therefore, in the three Merkel cell carcinoma cases with detectable retinoblastoma protein expression (Figure 3), phosphorylation of retinoblastoma protein is expected, given an intact retinoblastoma signaling pathway. However, in all eight cases, there was similar, minimal detectable phosphorylated-retinoblastoma protein, consistent with retinoblastoma dysregulation (Figure 4).

Fluorescence in situ hybridization confirms loss of RB1 in the only two cases of Merkel cell carcinoma as discovered by whole exome sequencing. (a) One case is Merkel cell polyomavirus-negative (case 21). (b) The other case is polyomavirus-positive (case 27). Normal RB1 copy number was observed in the other cases (not shown). Red=RB1; green=13q14.

Retinoblastoma protein immunoreactivity is absent in all Merkel cell carcinoma cases with a genetic mutation; and reactivity is present in all cases without a detected mutation. (a) Case 24—retinoblastoma-positive (no genetic mutation). (b) Case 06—retinoblastoma-positive (no genetic mutation). (c) Case 27—retinoblastoma-negative (RB1 copy number loss). (d) Case 39—retinoblastoma-positive (no genetic mutation). (e) Case 18—retinoblastoma-negative (deletion). (f) Case 33—retinoblastoma-negative (nonsense truncating mutation). (g) Case 29—retinoblastoma-negative (nonsense truncating mutation). (h) Case 21—retinoblastoma-negative (nonsense truncating mutation and RB1 copy number loss). Internal positive controls for retinoblastoma are seen in stromal and lymph node lymphocytes. All images were taken at × 200 original magnification.

Phosphorylated-retinoblastoma protein immunoreactivity is absent in all Merkel cell carcinoma cases, including those with pan-retinoblastoma staining. (a) Case 24, (b) Case 06, (c) Case 27, (d) Case 39, (e) Case 18, (f) Case 33, (g) Case 29, and (h) Case 21. Internal positive controls for phosphorylated-retinoblastoma are seen in few stromal and lymph node lymphocytes. All images were taken at × 200 original magnification.
Comparison of Variant Allele Fractions
To determine whether other gene variants were part of the same founder clone containing RB1 nonsense mutations, arising either before or at the time of the RB1 mutation, we examined the variant allele fraction of single-nucleotide variations as has been previously described.37 We first examined the allele fraction of single-nucleotide variations present in dbSNP (representing constitutional variants) to non-dbSNP variants (representing ‘personal’ constitutional variants and true somatically acquired mutations). As expected, although single-nucleotide variations present in dbSNP had allele fractions of 50 or 100% consistent with hetero- or homozygous constitutional variants, those not present in the dbSNP showed skewing of the allele fractions indicating the presence of true somatically acquired variants, present at variable allele fractions due to stromal cell dilution, copy number variation, tumor heterogeneity, and so on (Figure 5). RB1 variant allele fractions in the three cases (21, 29, and 33) with truncating retinoblastoma protein mutations ranged from 76.8 to 92.8%, with a mean of 85.2% (Figure 6). The increased allele fractions of RB1 truncating mutations to greater than 50% likely represent a homozygous mutation within tumor cells, which are diluted in a background of non-tumor cells. However, it is not possible to determine whether the RB1 mutations in each tumor represent a homozygous variant in a heterozygous background vs a homozygous acquired mutation. The number of genes with single-nucleotide variations occurring at an allele frequency ±5% within that of RB1, showed high variability between cases (Supplementary Table 1). This set included 10 genes for case 21, 31 genes for case 31, and 210 genes for case 29. None of the genes with single-nucleotide variations clustering around RB1 were recurrent among all three cases.

Variant allele fractions show two major populations, around 50% or 100% for the single-nucleotide polymorphisms (red, present in dbSNP), consistent with heterozygous or homozygous constitutional polymorphisms. The non-dbSNP variants (blue) occur mostly around 50%, likely indicative of ‘personal single-nucleotide polymorphisms’ (eg, variants not present in dbSNP). However, a number of the non-single-nucleotide polymorphism variants show substantial deviation from the expected 50 or 100% allelic fractions, indicative of true somatic variants or variants in regions of copy number variation. Merkel cell polyomavirus-positive cases: (a) Case 24, (b) Case 06, (c) Case 27, (d) Case 39, and (e) Case 18. Polyomavirus-negative cases: (f) Case 33, (g) Case 29, and (h) Case 21.

Combined allele frequency for all Merkel cell carcinoma cases. The frequency of retinoblastoma gene (RB1) non-single-nucleotide polymorphisms occurs around 85%. Other retinoblastoma pathway genes occur at lower allelic frequencies, including RBL1 (around 45%) and RBL2 (around 35%).
Comparison with Published Data Sets
To investigate whether the genetic signature of Merkel cell carcinoma is unique or whether it shows similarity to other cancer types, we compared the gene variants found in Merkel cell carcinoma to publicly available cancer-associated genetic mutations in the Catalog of Somatic Mutations in Cancer (COSMIC) database (Table 6). On average, 28 variants per case were present in COSMIC, with a range from 7 to 61. Only 13 of these mutations represented recurrent mutations, occurring in at least two Merkel cell carcinoma cases. Recurrently mutated variants occurred in AGBL2, ARMC4, FOXK1, LYZL2, SLC25A26, GLUD2, ZNRF4, PLEC, ZXDB, TP53, OR11G2, PCLO, and TTN. The most frequently mutated genes present in COSMIC were LYZL2, FOXK1, and SLC25A26, with each having mutations in four of eight cases. Overall, it does not appear that the gene mutations found in Merkel cell carcinoma show compelling overlap with other well-studied cancer types, suggesting a unique pathogenesis in Merkel cell carcinoma.
Discussion
For the first time, whole exome sequencing was used as an unbiased tool to characterize the genetic landscape of Merkel cell carcinoma, utilizing archival formalin-fixed paraffin-embedded tissue. This is in contrast to previous studies that have taken targeted approaches to define somatic mutations in Merkel cell carcinoma. These genes include PIK3CA, TP53, and PTEN.40, 41, 42, 43 In our study cases, PIK3CA and TP53, but not PTEN, were found to harbor mutations, but only in small number of cases (Table 5). Using the gene level recurrence as a metric, we identified the retinoblastoma pathway as critical to Merkel cell carcinoma pathogenesis. We acknowledge, however, that without concurrent exome data from paired normal tissue in each case, we cannot fully exclude that the ‘mutations’ identified represent benign polymorphisms not present in dbSNP; estimates of such ‘personal single-nucleotide variants’ are ∼200 coding region variants per normal individual and are therefore unlikely to account for all variants identified in each case.44 To further ameliorate this potential source of false discovery, we compared our data with the COSMIC database of known somatic mutations in cancer, looked only for recurrent mutations present in more than one case (and therefore unlikely to represent low prevalence polymorphisms), and examined the variant allele fraction of potential mutations.
Other groups have also provided evidence for retinoblastoma pathway dysregulation in subsets of Merkel cell carcinoma. Merkel cell polyomavirus has a large T antigen LXCXE domain that, when expressed, binds directly to retinoblastoma protein.45 Several lines of evidence have suggested that in polyomavirus-positive cases, retinoblastoma dysregulation occurs secondary to maintenance and expression of the large T antigen, and specifically the retinoblastoma protein-binding region of the large T antigen.12, 15, 16, 17, 18, 19, 20, 21 The large T antigen of integrated Merkel cell polyomavirus has been shown to have varying mutations among Merkel cell carcinoma cases, but the mutations invariably spare the retinoblastoma protein-binding domain.18 At the human genome level, prior studies by others have provided evidence of loss of RB1 in subsets of Merkel cell carcinoma. In 1997, Leonard and Hayward46 demonstrated a loss of heterozygosity of 13q14, the chromosomal region of RB1 locus, in 18/24 (75%) of Merkel cell carcinoma cases and additionally used western blot analysis to show that cell lines derived from 9/18 of these patients had an absence of detectable retinoblastoma protein. Later, array comparative genomic hybridization studies involving the RB1 locus by Van Gele et al47 and Paulson et al48 showed the deletion of 13q in 8/24 (33%) Merkel cell carcinoma cases and 13q14-13q21 in 6/23 (26%) Merkel cell carcinoma cases, respectively.
We found that polyomavirus-negative cases with little or no detectable polyomavirus by sensitive real-time polymerase chain reaction had truncating, nonsense RB1 mutations. Even though two of the five polyomavirus-positive cases showed RB1 deletions (one case with a deletion and one with copy number variation), there were no single-nucleotide variation truncating nonsense mutations within polyomavirus-positive cases. This suggests a unique genetic mechanism to RB1 inactivation occurring within polyomavirus-negative cases; however, given the small sample size in this study, we cannot exclude that such mutations may also be present in polyomavirus-positive cases. Further we note that, although two polyomavirus-negative cases had DNA-level mutations in RB1, one resulted in copy loss of RB1, and the other a 68 amino-acid deletion near the C-terminus, and may result in different functional effects than the RB1 truncating mutations seen in polyomavirus-negative cases.
Other groups have reported a similar correlation between retinoblastoma protein expression and Merkel cell polyomavirus copy number. Bhatia et al22 linked the presence of retinoblastoma protein by immunohistochemistry to cases of Merkel cell carcinoma with a polyomavirus load of at least 0.06 viral copies/cell (n=9), as detected by real-time polymerase chain reaction. Merkel cell carcinoma cases with polyomavirus viral loads ranging from 0 to 0.0035 viral copies/cell (n=14) had an absence of retinoblastoma protein. However, a study by Houben et al49 showed retinoblastoma protein expression by immunohistochemistry in every tested Merkel cell carcinoma case (n=50), including those with extremely low levels of viral copies/cell. It is possible that the discrepant findings were due to differing antibodies used or antigen retrieval techniques. A recent gene expression study by Harms et al50 comparing transcripts between Merkel cell polyomavirus-positive and polyomavirus-negative Merkel cell carcinoma cases showed that virus-negative cases have a relative 2.4-fold lower expression of RB1 and that retinoblastoma protein immunohistochemistry corresponded to Merkel cell polyomavirus status and RB1 transcript levels. In this study, we link the presence of decreased retinoblastoma protein expression to specific genetic mutations. Overall, these mutations tend to occur in Merkel cell carcinoma cases with low to no detectable polyomavirus arguing that retinoblastoma abrogation is required for Merkel cell carcinoma pathogenesis in the absence of polyomavirus.
Using an unbiased genomic approach, we identified the retinoblastoma pathway as critical in Merkel cell carcinoma pathogenesis and validated that retinoblastoma protein was decreased or absent in Merkel cell carcinoma cases with RB1 mutations. Further, we demonstrate that in Merkel cell carcinoma cases with intact retinoblastoma protein expression and without the evidence of RB1 mutations, the majority of retinoblastoma protein exists in the active, unphosphorylated form, despite frequent tumor cell division. This finding suggests retinoblastoma protein dysregulation by an alternative pathway in some cases of polyomavirus-positive Merkel cell carcinoma, such as direct large T antigen binding and subsequent non-phosphorylation-dependent inactivation of retinoblastoma protein. On the basis of these findings, we propose a model of Merkel cell carcinoma oncogenesis by which two separate pathways, a polyomavirus-dependent pathway in which retinoblastoma protein is functionally inactivated and a polyomavirus-independent pathway in which RB1 sustains somatic mutation, both of which require retinoblastoma protein dysregulation to produce an overlapping Merkel cell carcinoma phenotype (Figure 7). An alternative explanation for RB1 mutation in Merkel cell carcinoma might include the proposed model of hit-and-run oncogenesis in which Merkel cell polyomavirus integrates in to the host genome of all Merkel cell carcinoma cases, does genetic damage, either persists as clonally integrated virus or is expelled from the human genome by various repair mechanisms.51 Integrating our data with this model, Merkel cell polyomavirus may initiate Merkel cell carcinoma tumorogenesis in a subset of cases, including a genetic ‘hit’ to RB1 such as nonsense truncating mutations. After this ‘hit’ occurs, Merkel cell polyomavirus is no longer necessary for Merkel cell carcinoma progression and maintenance and the virus leaves (‘runs’ from) the human host genome. In any case, the retinoblastoma pathway appears to have an important role in Merkel cell carcinoma. Therapeutic targeting of the retinoblastoma pathway, specifically downstream of the retinoblastoma protein itself, by small-molecule inhibitors has been proposed in several tumor types with retinoblastoma protein loss (recently reviewed52). Perhaps future research focused on targeting the retinoblastoma pathway in Merkel cell carcinoma may offer clinical benefit for this highly aggressive cancer.

Proposed mechanism of Merkel cell carcinoma oncogenesis involving the retinoblastoma pathway. In this model, the retinoblastoma pathway is dysregulated in both Merkel cell polyomavirus (MCPyV)-positive and polyomavirus-negative cases, which leads to an indistinguishable morphological and clinical phenotype.
References
Calder KB, Smoller BR . New insights into merkel cell carcinoma. Adv Anat Pathol 2010;17:155–161.
Agelli M, Clegg LX, Becker JC et al. The etiology and epidemiology of merkel cell carcinoma. Curr Probl Cancer 2010;34:14–37.
Albores-Saavedra J, Batich K, Chable-Montero F et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 2010;37:20–27.
Lemos BD, Storer BE, Iyer JG et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol 2010;63:751–761.
Duncavage EJ, Zehnbauer BA, Pfeifer JD . Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol 2009;22:516–521.
Feng H, Shuda M, Chang Y et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319:1096–1100.
Sihto H, Kukko H, Koljonen V et al. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst 2009;101:938–945.
Chen T, Hedman L, Mattila PS et al. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J Clin Virol 2011;50:125–129.
Pastrana DV, Tolstov YL, Becker JC et al. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog 2009;5:e1000578.
Tolstov YL, Pastrana DV, Feng H et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer 2009;125:1250–1256.
Dalianis T, Hirsch HH . Human polyomaviruses in disease and cancer. Virology 2013;437:63–72.
Chang Y, Moore PS . Merkel cell carcinoma: a virus-induced human cancer. Annu Rev Pathol 2012;7:123–144.
Duncavage EJ, Magrini V, Becker N et al. Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2011;13:325–333.
Martel-Jantin C, Filippone C, Cassar O et al. Genetic variability and integration of Merkel cell polyomavirus in Merkel cell carcinoma. Virology 2012;426:134–142.
Angermeyer S, Hesbacher S, Becker JC et al. Merkel cell polyomavirus positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol 2013;133:2059–2064.
Cheng J, Rozenblatt-Rosen O, Paulson KG et al. Merkel cell polyomavirus large t antigen has growth promoting and inhibitory activities. J Virol 2013;87:6116–6126.
Demetriou SK, Ona-Vu K, Sullivan EM et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer 2012;131:1818–1827.
Houben R, Adam C, Baeurle A et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer 2012;130:847–856.
Houben R, Shuda M, Weinkam R et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol 2010;84:7064–7072.
Sastre-Garau X, Peter M, Avril MF et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol 2009;218:48–56.
Sihto H, Kukko H, Koljonen V et al. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res 2011;17:4806–4813.
Bhatia K, Goedert JJ, Modali R et al. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. Int J Cancer 2010;126:2240–2246.
Handschel J, Muller D, Depprich RA et al. The new polyomavirus (MCPyV) does not affect the clinical course in MCCs. Int J Oral Maxillofac Surg 2010;39:1086–1090.
Schrama D, Peitsch WK, Zapatka M et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol 2011;131:1631–1638.
Asioli S, Righi A, de Biase D et al. Expression of p63 is the sole independent marker of aggressiveness in localised (stage I-II) Merkel cell carcinomas. Mod Pathol 2011;24:1451–1461.
Kuwamoto S, Higaki H, Kanai K et al. Association of Merkel cell polyomavirus infection with morphologic differences in Merkel cell carcinoma. Hum Pathol 2011;42:632–640.
Teman CJ, Tripp SR, Perkins SL et al. Merkel cell polyomavirus (MCPyV) in chronic lymphocytic leukemia/small lymphocytic lymphoma. Leuk Res 2011;35:689–692.
Cimino PJ Jr., Bahler DW, Duncavage EJ . Detection of Merkel cell polyomavirus in chronic lymphocytic leukemia T-cells. Exp Mol Pathol 2013;94:40–44.
DePristo MA, Banks E, Poplin R et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
McKenna A, Hanna M, Banks E et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303.
Li H, Handsaker B, Wysoker A et al. The Sequence alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079.
Ye K, Schulz MH, Long Q et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009;25:2865–2871.
Chen K, Wallis JW, McLellan MD et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009;6:677–681.
Li J, Lupat R, Amarasinghe KC et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics 2012;28:1307–1313.
Bamford S, Dawson E, Forbes S et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004;91:355–358.
Forbes SA, Bindal N, Bamford S et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–D950.
Walter MJ, Shen D, Ding L et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 2012;366:1090–1098.
Higaki-Mori H, Kuwamoto S, Iwasaki T et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol 2012;43:2282–2291.
Adzhubei IA, Schmidt S, Peshkin L et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Hafner C, Houben R, Baeurle A et al. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS One 2012;7:e31255.
Lassacher A, Heitzer E, Kerl H et al. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J Invest Dermatol 2008;128:1788–1796.
Nardi V, Song Y, Santamaria-Barria JA et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res 2012;18:1227–1236.
Van Gele M, Leonard JH, Van Roy N et al. Frequent allelic loss at 10q23 but low incidence of PTEN mutations in Merkel cell carcinoma. Int J Cancer 2001;92:409–413.
Bamshad MJ, Ng SB, Bigham AW et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12:745–755.
Shuda M, Feng H, Kwun HJ et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA 2008;105:16272–16277.
Leonard JH, Hayard N . Loss of heterozygosity of chromosome 13 in Merkel cell carcinoma. Genes Chromosomes Cancer 1997;20:93–97.
Van Gele M, Speleman F, Vandesompele J et al. Characteristic pattern of chromosomal gains and losses in Merkel cell carcinoma detected by comparative genomic hybridization. Cancer Res 1998;58:1503–1508.
Paulson KG, Lemos BD, Feng B et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol 2009;129:1547–1555.
Houben R, Schrama D, Alb M et al. Comparable expression and phosphorylation of the retinoblastoma protein in Merkel cell polyoma virus-positive and negative Merkel cell carcinoma. Int J Cancer 2010;126:796–798.
Harms PW, Patel RM, Verhaegen ME et al. Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J Invest Dermatol 2013;133:936–945.
Houben R, Grimm J, Willmes C et al. Merkel cell carcinoma and Merkel cell polyomavirus: evidence for hit-and-run oncogenesis. J Invest Dermatol 2012;132:254–256.
Clem BF, Chesney J . Molecular pathways: regulation of metabolism by RB. Clin Cancer Res 2012;18:6096–6100.
Acknowledgements
We thank Xiaopei Zhu for assistance with DNA extraction. We also thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for assistance with DNA sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article
Cite this article
Cimino, P., Robirds, D., Tripp, S. et al. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Mod Pathol 27, 1073–1087 (2014). https://doi.org/10.1038/modpathol.2013.235
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.235
Keywords
This article is cited by
-
Investigation of the RB1-SOX2 axis constitutes a tool for viral status determination and diagnosis in Merkel cell carcinoma
Virchows Archiv (2022)
-
Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts
Scientific Reports (2020)
-
Invasive squamous cell carcinomas and precursor lesions on UV-exposed epithelia demonstrate concordant genomic complexity in driver genes
Modern Pathology (2020)
-
The biology and treatment of Merkel cell carcinoma: current understanding and research priorities
Nature Reviews Clinical Oncology (2018)
-
Merkel cell carcinoma
Nature Reviews Disease Primers (2017)
