
Abstract
Although benign hemangiomas are among the most common diagnoses amid connective tissue tumors, sarcomas showing endothelial differentiation (ie, angiosarcoma and epithelioid hemangioendothelioma) represent under 1% of all sarcoma diagnoses, and thus it is likely that fewer than 500 people in the United States are affected each year. Differential diagnosis of malignant vascular tumors can be often quite challenging, either at the low end of the spectrum, distinguishing an epithelioid hemangioendothelioma from an epithelioid hemangioma, or at the high-grade end of the spectrum, between an angiosarcoma and a malignant epithelioid hemangioendothelioma. Within this differential diagnosis both clinico-radiological features (ie, size and multifocality) and immunohistochemical markers (ie, expression of endothelial markers) are often similar and cannot distinguish between benign and malignant vascular lesions. Molecular ancillary tests have long been needed for a more objective diagnosis and classification of malignant vascular tumors, particularly within the epithelioid phenotype. As significant advances have been recently made in understanding the genetic signatures of vascular tumors, this review will take the opportunity to provide a detailed update on these findings. Specifically, this article will focus on the following aspects: (1) pathological and molecular features of epithelioid hemangioendothelioma, including the more common WWTR1–CAMTA1 fusion, as well as the recently described YAP1–TFE3 fusion, identified in a morphological variant of epithelioid hemangioendothelioma; (2) discuss the heterogeneity of angiosarcoma clinical, morphological and genetic spectrum, with particular emphasis of MYC and FLT4 gene amplification in radiation-induced angiosarcoma; and (3) provide a practical guide in the differential diagnosis of epithelioid vascular tumors using molecular testing.
Similar content being viewed by others

Epithelioid hemangioendothelioma
Incidence, Demographics. and Terminology
Epithelioid hemangioendothelioma (EHE) is a rare soft tissue tumor, substantially less common than angiosarcoma, but given the more indolent nature of the tumor it encompasses a larger prevalent population.1 It occurs in a younger population, with a peak incidence in the 4th to 5th decade.2 Women appear to be affected more commonly than men. In a survey of EHE patients from an EHE support group, overall survival was 73% at 5 years.3
EHE of soft tissue was first described by Weiss and Enzinger.4 As its clinical behavior is intermediate between a benign hemangioma and a high-grade angiosarcoma, the term ‘hemangioendothelioma’ was suggested. Subsequently, EHE of soft tissue was stratified into two risk groups of malignancy, classic and malignant EHE, based on mitotic activity and size, in an effort to reflect different biological subsets.5, 6
Pathological Features
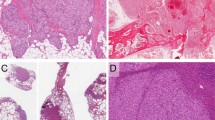
EHEs have a propensity for angiocentric growth, expanding the vessel wall, obliterating the lumen and spreading centrifugally into surrounding tissue where they induce a sclerotic response (Figures 1a-c). Microscopically, EHE are arranged in single files, cords and small nests (Figures 1d,e), typically lacking well-formed vascular channels, with only immature, intracytoplasmic lumina being observed (Figures 1f). Other distinctive features include an infiltrative growth pattern, encasing surrounding structures and lack of a lobular growth pattern typically seen with benign hemangiomas. EHE cells are separated by a characteristic extracellular sulfated acid-rich matrix, varying from a light blue (myxoid, myxo-chondroid, Figures 1f,g) to a deep pink (hyaline, Figure 1h) extracellular stroma. The cells have a pale to more densely eosinophilic cytoplasm containing vacuoles, which deform the cytoplasm, so-called ‘blister cells’. Some vacuoles contain fragmented erythrocytes. In most cases, the cells show a low nuclear grade, however, a small subset of cases show moderate to marked nuclear pleomorphism, with overt hyperchromasia and increased mitotic activity (Figure 1i). This latter appearance may be quite challenging to separate from angiosarcoma in a small biopsy sample.

Conventional epithelioid hemangioendothelioma, pathological findings: (a) gross appearance of an epithelioid hemangioendothelioma (EHE) of the superior vena cava, tumor being entirely confined to the vein wall; (b) van Gieson elastic stain highlighting the elastic structure of a vessel wall showing complete obliteration with tumor; (c) angiocentric growth involving a small vessel wall in a soft tissue EHE; microscopic appearance showing cords (d) or solid sheets (e) of epithelioid cells with moderate amount of pale eosinophilic cytoplasm; (f) the presence of intracytoplasmic lumens (blisters cells) is a common feature, as is the myxochodnroid quality of the extracellular stroma; occasionally the matrix may more myxoid (g) or hyalinized (h) in appearance. (i) Malignant EHE with hyperchromasia, nuclear pleomorphism and increased mitotic activity.
Genetics
A t(1;3)(p36.3;q25) translocation was initially reported in two cases of EHE, suggesting that this may represent a recurrent abnormality in this subgroup of tumors.7 Ten years later, two back-to-back studies, using different methodologies, were able to clone the genes involved in the t(1;3)(p36.3;q25) translocation.8, 9 WWTR1 (also called TAZ), on 3q25, is a gene involved in 14-3-3 transcriptional factor activation and signaling in the hippo pathway that is normally highly expressed in endothelial cells.10 WWTR1 fuses to CAMTA1, on 1p36, which belongs to a family of calmodulin-binding transcription activators, normally found only in brain. The 1p36 region is frequently lost in neuroblastoma and gliomas, implicating CAMTA1 as a tumor suppressor gene.11, 12 Most cases with classic morphology (ie, lacking mature vascular channel formation) examined so far contain this translocation,8, 9 which is not directly targetable with present day therapeutics, but may reveal downstream targets in future studies.
In 50% of cases, EHE have a multifocal clinical presentation.3 It has long been debated if this occurrence is truly multifocal or represents an unusual pattern of loco-regional metastasis. This is particularly relevant in visceral EHE, where the multifocal disease is limited to one organ (ie, liver, lung), without dissemination to other distant sites. As a result of this clinical behavior, patients with multifocal liver EHE are candidates for liver transplantation. Thus, by analysis of WWTR1–CAMTA1 breakpoints in multiple nodules from two patients with multifocal hepatic EHE, we were able to prove recently that this phenomenon is monoclonal, representing metastatic implants of the same neoplastic clone rather than a ‘field-effect’ or synchronous occurrence of multiple neoplastic clones.13
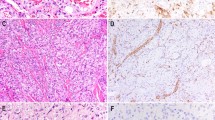
In a recent study, we identified the presence of YAP1–TFE3 gene fusion in a specific subset of EHE, lacking the WWTR1–CAMTA1 fusion and displaying a somewhat distinct morphology and strong TFE3 expression by immunohistochemistry.14 YAP1 transcriptional co-activator is a major downstream effector of the Hippo pathway and shares significant functional and sequence homology with WWTR1. As the S127 14-3-3 binding site, WW domain and its C-terminal transactivation domain are not retained in the fusion, it is most likely that YAP1 provides a strong promoter to the oncogenic TFE3 function of the fusion protein.14 The anatomic distribution of these lesions is mainly within soft tissue, ranging from head and neck, trunk and extremities, with only one case occurring in the bone (vertebral body). Additional cases were seen in the lung, with multifocal presentation.The tumors occurred in young adults, with a mean age of 30 years old, with no gender predilection. Microscopically, the tumors had abundant eosinophilic cytoplasm and well-developed vasoformative channels in most cases, compared with the classic EHE harboring WWTR1–CAMTA1 fusion. However, a morphological overlap between the two genetically distinct subsets of EHE was noted in few cases, with predominance of solid or nested-like growth pattern (Figure 2d). The tumors expressed endothelial markers (CD31 and ERG, Figure 2e), as well as strong nuclear TFE3 (Figure 2f), but were negative for cytokeratin and HMB45. In our series, the 10 patients followed an indolent clinical course, despite a high propensity for metastasis. In summary, the morphological hallmark for these tumors appeared to be the voluminous eosinophilic cytoplasm with mild-to-moderate cytological atypia and distinct vasoformative features. Larger studies are required to establish if TFE3-rearranged EHE have a distinct clinical behavior, compared with the more common EHE variant.

YAP1–TFE3 fusion positive epithelioid hemangioendothelioma display distinct morphological features including well-developed vasoformative growth and more abundant eosinophilic cytoplasm (a, b); occasionally showing overlapping features with conventional epithelioid hemangioendothelioma (EHE), such as cord-like growth pattern (c); (d) solid growth is variably seen. Immunohistochemical stains show diffuse and strong reactivity for vascular markers, such as CD31 (e), and TFE3 (f).
Prognostic Factors
The majority of soft tissue EHEs are indolent. However, 20–30% of tumors metastasize, and about 15% of patients die of their disease.4, 15 This contrasts with the behavior of most angiosarcomas. Overtly malignant lesions typically pursue an aggressive course. A combination of mitotic activity and tumor size have been used to stratify tumors into low and high-risk groups: patients with tumors >3 cm in diameter and having more than three mitoses/50 HPFs have a 5-year disease-specific survival of 59% in contrast to 100% survival in patients whose tumors lacked these features.5 In their study, however, the presence of nuclear pleomorphism and necrosis were not indicative of adverse outcome. Furthermore, these criteria do not apply for multifocal presentation and visceral locations. Thus, larger studies are needed to better define prognostic criteria in molecularly defined EHE patients.
Angiosarcoma
Incidence and Demographics
Angiosarcoma may arise in any part of the body, but is more common in soft tissue than in bone. The peak age of incidence appears to be the 7th decade, and men are affected more than women. The head and neck area is probably the most common site of diagnosis, and the most common site of radiation-induced angiosarcoma development is the breast. After rhabdomyosarcoma, angiosarcoma is probably the second most common sarcoma to arise from germ cell tumors.16
Pathogenesis
The most common cause of angiosarcoma appears to be therapeutic radiation, which was a well-recognized cause of hepatic angiosarcoma in the era when the thorium containing contrast agent Thorotrast was employed.17 Presently, the breast is the most common anatomic site affected by radiation-induced angiosarcoma.18
Angiosarcomas may arise after exposure to vinyl chloride, although they remain rare tumors even in an exposed population.19 Angiosarcomas are also observed after lymphedema from any cause, be it surgical, filarial or congenital, and defined as Stewart–Treves syndrome.20
Pathological Features
Soft tissue angiosarcomas are multinodular hemorrhagic masses often with secondary cystic degeneration and necrosis. They display a wide spectrum of morphological appearances, ranging from areas of well-formed, anastomosing vessels (Figure 3a) to solid sheets of high-grade epithelioid or spindled cells without clear vasoformation (Figures 3b,c). Multiple patterns may be present in the same tumor. Vasoformative areas consist of ramifying channels lined by atypical endothelial cells forming intraluminal buds and papillations. Solid areas lacking vasoformation are composed of high-grade spindled and epithelioid cells with abundant amphophilic to lightly eosinophilic cytoplasm, large vesicular nuclei and prominent nucleoli. Tumors in which these epithelioid cells predominate are classified as ‘epithelioid angiosarcomas.’ They are commonly confused with carcinomas because of morphological and immunophenotypic similarities.21, 22 The vast majority of angiosarcomas of soft tissue are high-grade neoplasms with brisk mitotic activity, coagulative necrosis and significant nuclear atypia. Extensive hemorrhage is commonly present and may suggest a hematoma. Careful sampling may be necessary to document malignant cells.

Angiosarcoma morphological spectrum includes vasoformative features and high-nuclear grade cytology, with open chromatin and prominent nucleoli in a hemorrhagic background (a), as well as undifferentiated histology lacking vascular channel formation and displaying either a predominant epithelioid (b) or spindle cell (c) phenotype. Comparative view of a post-radiation atypical vascular lesion (AVL) (d) and a radiation-induced angiosarcoma of the breast (e). Both lesions show an ill-defined growth, with more overt cytological atypia in the angiosarcoma (e); FISH for MYC gene abnormalities is very useful, showing high level of MYC amplification (red probe) as well as FLT4 (green probe) in radiation-induced angiosarcoma (f), but not in AVL (not shown). Angiosarcoma of the head and neck (scalp, sun-exposed) showing a ill-defined vascular proliferation obscured by a brisk lymphocytic infiltrate (g); primary breast angiosarcoma (grade I/III, according to Rosen 3-tier system) showing an infiltrative growth within the fat (h) and only mild cytological atypia (i).
Angiosarcomas express the typical vascular markers: CD34, CD31, Fli1, ERG and occasionally podoplanin (D2-40), a lymphatic marker.23, 24, 25, 26 Given differences in sensitivity and specificity, a panel of antibodies should be used.23, 24, 25 Some angiosarcomas co-express epithelial antigens (EMA, Cam5.2 and AE1/3), a fact that should be considered in distinguishing them from carcinoma. Immunostaining for the Kaposi sarcoma herpes virus is negative.
Genetics
By gene expression profiling angiosarcomas show distinct upregulation of vascular-specific receptor tyrosine kinases, including TIE1, KDR, TEK and FLT1, compared with other sarcoma types.27 In a subset (10%) of tumors KDR mutations (encoding for VEGFR2) were identified, which correlates with strong KDR protein expression and breast anatomic location regardless of exposure to radiation.27 The hot spot mutations were dispersed among extracellular, transmembrane and kinase domains of KDR. Transient transfection of the KDR mutants showed ligand-independent activation of the kinase, which was inhibited by specific KDR inhibitors, such as sunitinib and sorafenib.27
Furthermore, high level MYC amplification on 8q24 is a consistent hallmark of radiation-induced and lymphedema-associated angiosarcoma.28, 29 FLT4 (encoding for VEGFR3) co-amplification on 5q35 is detected in 25% of secondary angiosarcoma. Both MYC and FLT4 gene abnormalities have not been reported in radiation-associated atypical vascular lesions, thus far and can serve as a powerful molecular or immunohistochemical test in difficult cases (Figures 3d-f).29, 30, 31 The most common pattern of amplification for both MYC and FLT4 is present as large, confluent, usually single regions rather than individual small signals, known as ‘homogeneous staining regions’(Figure 3f). A small subset of primary angiosarcoma (ie, breast and bone) have also shown to carry MYC gene amplification.32 In our experience, MYC amplification was not detected in radiation-induced sarcomas that do not show an angiosarcoma phenotype.29
MYC has a crucial role in growth control, differentiation and apoptosis, and its aberrant expression is associated with several cancers. More recently, MYC was also implicated as having a major contribution in tumor angiogenesis,33, 34 by upregulating one of its direct targets, the miR-17–92 cluster. The predicted targets of miR-17–92 cluster are thrombospondin-1 (THBS1), encoding a potent endogenous inhibitor of angiogenesis, and connective tissue growth factor (CTGF), encoding an extracellular matrix-associated molecule involved in angiogenesis and metastatic progression.34 Indeed, a significant upregulation of miR-17–92 cluster is present in MYC-amplified angiosarcoma compared with angiosarcoma lacking MYC amplification and other vascular lesions.32 The mRNA expression of both THBS1 and CTGF was found to be significantly downregulated in the MYC-amplified angiosarcoma compared with other subsets.32 Thus, MYC amplification has a crucial role in the angiogenic phenotype of angiosarcoma through upregulation of the miR-17–-92 cluster, which subsequently downregulates thrombospondin-1 (THBS1), a potent endogenous inhibitor of angiogenesis.32
In contrast to other sarcomas with complex genomics, angiosarcoma shows a very low level of alterations in the p53 and PIK3CA/AKT/mTOR pathways. Specifically, no PTEN alterations were identified in a large series of angiosarcoma samples, including both primary and secondary cases.35 Furthermore, p53 mutations were detected in only 4% of cases, despite P53 overexpression by immunohistochemistry in 49% of tumors. Surprisingly, the P53 protein overexpression correlated with an inferior disease-free survival. Phosphorylated ribosomal protein S6 kinase (p-S6K) and/or phosphorylated eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1) overexpression was noted in 42% of patients, suggesting frequent activation of the PIK3CA/AKT/mTOR pathway.35 No BRAF or NRAS hot spot mutations were detected.
Site-Specific Pathological and Genetic Findings
Head and Neck Angiosarcoma represents the most prevalent subset and has a predilection for the sun-exposed skin of the scalp in older individuals. Their morphological appearance is somewhat distinct from other locations by its common association with a heavy lymphocytic infiltrate that can obscure its findings (Figure 3g). The lesions are typically composed of deceptive but highly infiltrative vascular proliferation, lined by relatively uniform cells, with scant cytoplasm and small but hyperchromatic nuclei. Due to its subtle cytological features of malignancy as well as dense inflammatory infiltrate an overt diagnosis of angiosarcoma may require repeated core biopsies. No specific molecular abnormalities have been so far associated with this clinico-pathological subset.
Primary breast angiosarcoma represents a unique subtype, occurring in younger women (peak in the 3rd and 4th decade of life), deep within the breast parenchyma. Not infrequently the lesions are quite well-differentiated, being composed of small to medium sized vascular channels, lined by flattened and relatively bland endothelial cells (Figure 3h,i). In some cases, the morphology is highly reminiscent of a hemangioma and a definitive distinction may not be possible in small core biopsies. The most important distinguishing factor is the diffuse infiltrative growth pattern within the breast parenchyma, which can help excluding a benign vascular neoplasm in a larger biopsy/lumpectomy. Primary breast angiosarcoma is also the only subtype for which histological grading (three-tier grading scheme based on the degree of vascular formation and cytological atypia) has been shown to correlate with outcome.36, 37 However, this concept has been recently challenged by Nascimento et al38, who found no correlation between histological grade and survival. Larger studies, including patients homogeneously treated at a single institution, are required to elucidate the role of histological grading in this subset of angiosarcomas. About 10% of cases show activating mutations in the KDR gene (encoding for VEGFR2).27
Radiation-induced and lymphedema-associated angiosarcoma are cutaneous lesions, which occur either in the radiation field (mostly radiotherapy for breast cancer) or in areas exposed to chronic lymphedema (mostly secondary to breast cancer). The patients typically present with multiple, ill-defined hemorrhagic plaques or firm nodules on the skin. Microscopically, the lesions involve the dermis and in locally advanced cases extend to underlying soft tissue and/or residual breast parenchyma. The tumors have an infiltrative growth and a variable degree of vasoformative features. Quite often tumors show a predominantly solid pattern, with well-formed vascular channels being a more focal finding. The lesional cells are round to epithelioid, with ill-defined cell borders and scant amphophilic cytoplasm and enlarged nuclei with either densely hyperchromatic or vesicular chromatin with prominent nucleoli. The morphology of the lymphedema-associated angiosarcoma (Stewart–Treves syndrome) recapitulates very closely the features described above for radiation-associated disease. Most angiosarcomas (>90%) in this group show high level of MYC amplification and in about 25% co-amplification of FLT4 (encoding for VEGFR3), which can be readily detected by FISH.29
Prognostic Factors
Soft tissue angiosarcomas are aggressive malignancies with a high rate of tumor-related deaths. Over half die within the first year.39 Features that are associated with poor outcome include older age, retroperitoneal location, large size and high Ki-67 values.39, 40
Differential diagnosis of epithelioid vascular tumors
Histologically, epithelioid vascular tumors show remarkable similarities, and diagnostic criteria to differentiate them still involve significant subjectivity. Particularly problematic has been the wide recognition of epithelioid hemangioma within bone, which historically has been lumped under the ‘hemangioendothelioma’ group of tumors, and only recently has been accepted as a distinct entity from EHE.41 Diagnostic challenges exist at the malignant end of the spectrum as well, the distinction between the so-called malignant EHE and high-grade epithelioid angiosarcoma can be quite arbitrary in a small sample material or when the angiosarcoma lacks vasoformative properties. Clinical behavior and, consequently, treatment and prognosis vary significantly among vascular tumors; thus, it is paramount to effectively distinguish them from each other.
The significant morphological overlap between these entities across the benign to malignant spectrum of epithelioid vascular neoplasms underscores the need for objective molecular markers to assist in an accurate classification. The presence of WWTR1–CAMTA1 fusion, typical for EHE, has not been detected in any of the morphological mimics, such as epithelioid hemangioma, pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma) or epithelioid angiosarcoma, and thus can serve as a useful molecular diagnostic tool in challenging cases. The presence of TFE3 immunoexpression and/or TFE3 gene rearrangement by FISH can confirm an unusual form of EHE with mature vessel formation from an epithelioid hemangioma.
Epithelioid Hemangioma
Epithelioid hemangioma is associated with a lobulated or multinodular growth, with a well-circumscribed or pushing border, showing overt vasoformative properties, with ‘mature’ vessels containing open lumina. The lesional cells show abundant, densely eosinophilic cytoplasm and may occasionally reveal the so-called ‘tomb-stone’ appearance. In some tumors, however, the cytoplasm has a more fine, feathery or foamy appearance, resembling that of histiocytic cells. Intracytoplasmic vacuoles may be present but typically not as a predominant feature. The nuclei may be enlarged with irregular nuclear contours and occasional grooves, but the cytological atypia is typically mild, lacking hyperchromasia or macro-nucleoli. Diagnostic pitfalls typically occur due to its under-recognized morphological spectrum, including solid growth, paucity or lack of eosinophils, the presence of mixed spindle and epithelioid growth patterns, as well as mild cytological atypia. The presence of spindling within an epithelioid hemangioma was not well recognized early on. Tumors displaying either a spindle or mixed epithelioid and spindle cell morphology were interpreted instead as a distinct variant of hemangioma, so-called ‘hemorrhagic epithelioid and spindle cell hemangioma’.42 This benign tumor, with propensity for multifocal presentation and acral bone involvement, showed some distinctive features from the typical epithelioid hemangioma, including abundant extravasated red blood cells and an admixture of epithelioid cells with bland spindle cells arranged in short fascicles. The authors have later acknowledged that this lesion represents in fact a histological variant of epithelioid hemangioma.43 Moreover, the presence of eosinophilic stromal infiltrate is only occasionally seen in the deep-seated soft tissue or bone epithelioid hemangioma, in contrast with their cutaneous counterpart.
Further confusion may occur upon review of the radiographic findings, which often show a locally aggressive and/or multifocal presentation, suggestive of malignancy.44 In fact, the radiological appearance of epithelioid hemangioma is not specific, having significant overlap with malignant vascular lesions, such as EHE or angiosarcoma.
Rare cases of epithelioid hemangiomas have been reported to carry TEK (TIE2) mutations; however, we were not able to detect mutations in the TIE2 hot-spots in 20 cases (unpublished, Cristina Antonescu personal communications). Particularly, important is the lack of CAMTA1, WWTR1 and TFE3 gene rearrangements in epithelioid hemangioma, which can be used to exclude an EHE diagnosis in challenging cases.
Pseudomyogenic Hemangioendothelioma (Epithelioid Sarcoma-like Hemangioendothelioma)
Epithelioid sarcoma-like hemangioendothelioma was initially described by Billings et al,45 as showing morphological features more in keeping with an epithelioid sarcoma, composed of epithelioid cells with dense, eosinophilic cytoplasm, but expressing endothelial markers, such as CD31 or Fli1, in addition to epithelial markers. The clinical features reported included multifocal presentation in the dermis and subcutis of the limbs of young adults, and associated with rare loco-regional metastases.
More recently, Hornick et al46 expanded the histological spectrum of this entity, showing that a significant number of these lesions show in fact a strikingly rhabdomyoblast-like appearance, with a brightly eosinophilic cytoplasm. Based on their findings, the term ‘pseudomyogenic hemangioendothelioma’ was preferred to better reflect the myoid-appearing spindle cell morphology. The lesions showed a distinctive, often multicentric presentation, in different tissue planes of a limb. The clinical presentation is indolent with a very low rate of loco-regional metastases. The tumors showed diffuse positivity for AE1:AE3 and Fli1, but variable reactivity for CD31. One of their cases was found to harbor a t(7;19) translocation.47
References
Brennan MF, Antonescu C, MAKI RG . Management of Soft Tissue Sarcoma. Springer: New York, NY, USA, 2013.
Miettinen M . Modern Soft Tissue Pathology: Tumors and Non-Neoplastic Conditions 1st edn. Cambridge University Press: Cambridge, UK, 2010.
Lau K, Massad M, Pollak C et al. Clinical patterns and outcome in epithelioid hemangioendothelioma with or without pulmonary involvement: insights from an internet registry in the study of a rare cancer. Chest 2011;140:1312–1318.
Weiss SW, Enzinger FM . Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer 1982;50:970–981.
Deyrup AT, Tighiouart M, Montag AG et al. Epithelioid hemangioendothelioma of soft tissue: a proposal for risk stratification based on 49 cases. Am J Surg Pathol 2008;32:924–927.
Fletcher CD, Unni K, Mertens F . WHO. Pathology and Genetics. Tumours of Soft Tissue and Bone. IARC Press: Lyon, 2002.
Mendlick MR, Nelson M, Pickering D et al. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am J Surg Pathol 2001;25:684–687.
Tanas MR, Sboner A, Oliveira AM et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med 2011;3:98ra82.
Errani C, Zhang L, Sung YS et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011;50:644–653.
Chan SW, Lim CJ, Chen L et al. The Hippo pathway in biological control and cancer development. J Cell Physiol 2011;226:928–939.
Attiyeh EF, London WB, Mosse YP et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 2005;353:2243–2253.
Barbashina V, Salazar P, Holland EC et al. Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res 2005;11:1119–1128.
Errani C, Sung YS, Zhang L et al. Monoclonality of multifocal epithelioid hemangioendothelioma of the liver by analysis of WWTR1-CAMTA1 breakpoints. Cancer Genet 2012;205:12–17.
Antonescu CR, Le Loarer F, Mosquera JM et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013;52:775–784.
Mentzel T, Beham A, Calonje E et al. Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 1997;21:363–374.
Contreras AL, Punar M, Tamboli P et al. Mediastinal germ cell tumors with an angiosarcomatous component: a report of 12 cases. Hum Pathol 2010;41:832–837.
Lipshutz GS, Brennan TV, Warren RS . Thorotrast-induced liver neoplasia: a collective review. J Am Coll Surg 2002;195:713–718.
Buchholz TA, Theriault RL, Niland JC et al. The use of radiation as a component of breast conservation therapy in National Comprehensive Cancer Network Centers. J Clin Oncol 2006;24:361–369.
Marion MJ, Boivin-Angele S . Vinyl chloride-specific mutations in humans and animals. IARC Sci Publ 1999;315–324.
Stewart FW, Treves N . Lymphangiosarcoma in postmastectomy lymphedema; a report of six cases in elephantiasis chirurgica. Cancer 1948;1:64–81.
Al-Abbadi MA, Almasri NM, Al-Quran S et al. Cytokeratin and epithelial membrane antigen expression in angiosarcomas: an immunohistochemical study of 33 cases. Arch Pathol Lab Med 2007;131:288–292.
Miettinen M, Fetsch JF . Distribution of keratins in normal endothelial cells and a spectrum of vascular tumors: implications in tumor diagnosis. Hum Pathol 2000;31:1062–1067.
Folpe AL, Chand EM, Goldblum JR et al. Expression of Fli-1, a nuclear transcription factor, distinguishes vascular neoplasms from potential mimics. Am J Surg Pathol 2001;25:1061–1066.
DeYoung BR, Swanson PE, Argenyi ZB et al. CD31 immunoreactivity in mesenchymal neoplasms of the skin and subcutis: report of 145 cases and review of putative immunohistologic markers of endothelial differentiation. J Cutan Pathol 1995;22:215–222.
Miettinen M, Lindenmayer AE, Chaubal A . Endothelial cell markers CD31, CD34, and BNH9 antibody to H- and Y-antigens--evaluation of their specificity and sensitivity in the diagnosis of vascular tumors and comparison with von Willebrand factor. Mod Pathol 1994;7:82–90.
Miettinen M, Wang ZF, Paetau A et al. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. Am J Surg Pathol 2011;35:432–441.
Antonescu CR, Yoshida A, Guo T et al. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res 2009;69:7175–7179.
Manner J, Radlwimmer B, Hohenberger P et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol 2010;176:34–39.
Guo T, Chang NE, Singer S et al. Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions. Genes Chromosomes Cancer 2011;50:25–33.
Fernandez AP, Sun Y, Tubbs RR et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol 2012;39:234–242.
Mentzel T, Schildhaus HU, Palmedo G et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol 2012;25:75–85.
Italiano A, Thomas R, Breen M et al. The miR-17-92 cluster and its target THBS1 are differentially expressed in angiosarcomas dependent on MYC amplification. Genes Chromosomes Cancer 2012;51:569–578.
Baudino TA, McKay C, Pendeville-Samain H et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 2002;16:2530–2543.
Dews M, Homayouni A, Yu D et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 2006;38:1060–1065.
Italiano A, Chen CL, Thomas R et al. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: a pattern distinct from other sarcomas with complex genomics. Cancer 2012;118:5878–5887.
Donnell RM, Rosen PP, Lieberman PH et al. Angiosarcoma and other vascular tumors of the breast. Am J Surg Pathol 1981;5:629–642.
Rosen PP, Kimmel M, Ernsberger D . Mammary angiosarcoma. The prognostic significance of tumor differentiation. Cancer 1988;62:2145–2151.
Nascimento AF, Raut CP, Fletcher CD . Primary angiosarcoma of the breast: clinicopathologic analysis of 49 cases, suggesting that grade is not prognostic. Am J Surg Pathol 2008;32:1896–1904.
Meis-Kindblom JM, Kindblom LG . Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol 1998;22:683–697.
Fayette J, Martin E, Piperno-Neumann S et al. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: a retrospective study of 161 cases. Ann Oncol 2007;18:2030–2036.
O’Connell JX, Nielsen GP, Rosenberg AE . Epithelioid vascular tumors of bone: a review and proposal of a classification scheme. Adv Anat Pathol 2001;8:74–82.
Keel SB, Rosenberg AE . Hemorrhagic epithelioid and spindle cell hemangioma: a newly recognized, unique vascular tumor of bone. Cancer 1999;85:1966–1972.
Nielsen GP, Srivastava A, Kattapuram S et al. Epithelioid hemangioma of bone revisited: a study of 50 cases. Am J Surg Pathol 2009;33:270–277.
Errani C, Zhang L, Panicek DM et al. Epithelioid hemangioma of bone and soft tissue: a reappraisal of a controversial entity. Clin Orthop Relat Res 2012;470:1498–1506.
Billings SD, Folpe AL, Weiss SW . Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol 2003;27:48–57.
Hornick JL, Fletcher CD . Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol 2011;35:190–201.
Trombetta D, Magnusson L, von Steyern FV et al. Translocation t(7;19)(q22;q13)-a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet 2011;204:211–215.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
About this article
Cite this article
Antonescu, C. Malignant vascular tumors—an update. Mod Pathol 27 (Suppl 1), S30–S38 (2014). https://doi.org/10.1038/modpathol.2013.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.176
Keywords
This article is cited by
-
Histological and immunohistochemical prognostic factors of primary angiosarcoma
Virchows Archiv (2023)
-
Epithelioid Hemangioendothelioma of the Mastoid Bone with Extension of Middle Cranial Fossa: A Case Report
Indian Journal of Otolaryngology and Head & Neck Surgery (2023)
-
Subcutaneous axillary primary epithelioid hemangioendothelioma: report of a rare case
Surgical Case Reports (2022)
-
A photoacoustic patch for three-dimensional imaging of hemoglobin and core temperature
Nature Communications (2022)
-
Neonatal vascular anomalies manifesting as soft-tissue masses
Pediatric Radiology (2022)
