
Abstract
B acute lymphoblastic leukemia (B-ALL) with t(14;19)(q32;p13.1), in which IGH and EPOR are juxtaposed, has been reported rarely. We describe the clinicopathological features of six patients, three men and three women, with a median age of 39 years. Initial and follow-up bone marrow samples were examined from each patient. The clinical, morphologic, and immunophenotypic results were compared with data obtained from conventional cytogenetic analysis and by using home-brew fluorescence in situ hybridization (FISH) probes for IGH at 14q32 and EPOR at 19p13.1. The bone marrow specimens were hypercellular (median 90%; range 80–100%), with a median blast count of 90% (range 60–93%). Immunophenotypic analysis performed by flow cytometry demonstrated a stable, precursor B-cell immunophenotype. The t(14;19)(q32;p13.1) was present in all cases with morphologic evidence of disease. The translocation was stable and appeared morphologically subtle on conventional karyotypic analysis. Detection was facilitated using FISH, which confirmed IGH/EPOR rearrangement in all cases. All patients received aggressive multiagent chemotherapy as part of a variety of regimens. Four of six patients achieved an initial complete remission, but all relapsed. At last follow-up, five of six patients had died of disease (median survival, 12 months after diagnosis). We conclude that B-ALL associated with t(14;19)(q32;p13.1) is a distinctive form of disease that is associated with younger patient age and an aggressive clinical course.
Similar content being viewed by others
Main
B acute lymphoblastic leukemia (B-ALL) is a neoplasm of precursor B cells (lymphoblasts) that primarily involves the bone marrow and blood. B-ALL encompasses many subsets that share overlapping clinical, morphologic, and immunophenotypic features, but are genetically distinct. The World Health Organization classification of hematopoietic and lymphoid neoplasms currently recognizes eight subtypes of B-ALL, seven of which are defined by the presence of specific genetic abnormalities.1 These genetic abnormalities range from numerical and structural changes to balanced translocations and impart distinct clinicopathologic features.
Reciprocal translocations that juxtapose a potential oncogene with regulatory elements of the IGH locus on chromosome 14q32 are common in mature B-cell neoplasms, but are uncommon in B-ALL, occurring in <5% of all cases.2, 3 However, the increased availability of commercial IGH probes, along with advances in molecular and cytogenetic techniques, has led to the identification of several recurrent translocations involving IGH in B-ALL.3 These translocations result in deregulated expression of the partner gene, presumably because of the resulting proximity to enhancer elements within the IGH locus.4 Recently, a small number of cases of B-ALL involving IGH and the erythropoietin receptor gene (EPOR) at 19p13.1 have been reported.5, 6 EPOR encodes a type 1 cytokine receptor involved in kinase signaling that is normally found on the surface of erythroid progenitor cells and is required for normal erythropoiesis.5, 7 B-ALL with genetic alterations involving cytokine and receptor signaling has been associated with an aggressive clinical course.6 However, the clinicopathologic features of B-ALL with t(14;19)(q32;p13.1) remain incompletely described.
In this report, we describe the clinicopathologic and cytogenetic features of six cases of B-ALL with t(14;19)(q32;p13.1)/IGH-EPOR, the largest series reported to date.
Materials and methods
Case Selection
We searched the database of the Clinical Cytogenetics Laboratory at the University of Texas MD Anderson Cancer Center from January 2007 to January 2013 and identified six patients with B-ALL associated with t(14;19)(q32;p13.1; Table 1). All patients were Philadelphia chromosome-negative by fluorescence in situ hybridization (FISH) and/or RT-PCR analysis. These cases represented <1% of all Philadelphia chromosome-negative B-ALL in our institution during the study interval. Clinical and laboratory data were obtained by review of the medical records. All patient information was gathered in accordance with an internal review board-approved protocol.
Morphologic Examination
For each case, we reviewed peripheral blood and bone marrow aspirate smears, touch imprints, aspirate clot sections, and core biopsy specimens. Peripheral blood smears were stained with May–Grünwald–Giemsa stain, and a manual 100-cell differential white blood cell count was performed. Differential 500-cell counts were performed on bone marrow aspirate smears or touch imprints stained with Wright–Giemsa stain. Samples from patients presenting to our institution with relapsed/refractory disease were compared with the original, diagnostic bone marrow specimens from the referring institution.
Immunophenotypic Analysis
Immunophenotypic analysis was performed by flow cytometry using bone marrow aspirate samples and a FACScan instrument (Becton-Dickinson Biosciences, San Jose, CA, USA) as described previously.8 Samples were first evaluated using an acute leukemia panel, followed by an extended B-ALL panel. Follow-up samples were examined using a minimal residual disease panel for B-ALL, as described previously.9
Conventional Cytogenetic and FISH Analyses
Conventional cytogenetic analysis was performed on metaphase cells prepared from bone marrow aspirate specimens that were cultured for 24 h without mitogens. We analyzed 20 Giemsa-banded metaphases and reported the results using the International System for Human Cytogenetic Nomenclature (2009, 2013).
FISH analysis for BCR-ABL1 was performed on interphase nuclei using the Vysis LSI BCR/ABL ES dual color translocation probe (Abbott Molecular, Des Plaines, IL, USA). A minimum of 500 interphase nuclei were analyzed for each case.
We constructed FISH probes for IGH and EPOR from bacterial artificial chromosome clones obtained from a commercial vendor (Invitrogen/Life Technologies, Grand Island, NY, USA). The clones were cultured in LB media, and DNA was isolated using a PureLink HiPure Plasmid DNA Purification kit (Invitrogen/Life Technologies). The DNA was labeled with SpectrumGreen-dUTP for IGH and with SpectrumRed-dUTP for EPOR by nick translation (Abbott Molecular, Downers Grove, IL, USA). The probes were mapped to normal metaphases to confirm the location of IGH at chromosome 14q32 and EPOR at 19p13.1. The IGH/EPOR rearrangement was demonstrated using fluorescence microscopy and appeared as a single yellow fusion signal on the der(14)t(14;19)(q32;p13.1). A value of ≥2.8% was considered a positive result. A minimum of 200 interphase nuclei obtained from the bone marrow cultures were analyzed for each case. All specimens obtained at our institution throughout the disease course were assessed by both conventional cytogenetic and FISH analyses for t(14;19).
Quantitative Real-Time PCR Assay
The presence and amount of BCR-ABL1 fusion transcripts was assessed using a multiplex quantitative real-time PCR assay that simultaneously detects b2a2, b3a2, and e1a2 transcripts.10
Survival Analysis
Response to therapy, time to relapse, and overall survival were calculated for all patients. Complete remission was defined as <5% leukemic blasts in the bone marrow and peripheral blood as determined by morphologic review and flow cytometric immunophenotyping. Time to relapse was calculated as the time between first in-hospital sample with complete remission and first in-hospital sample with morphologic and immunophenotypic evidence of ≥5% leukemic blasts. Overall survival was calculated using GraphPad Prism Software (Version 6.01, La Jolla, CA, USA) from the date of first available diagnostic bone marrow sample to the date of death from any cause. An additional time to relapse and overall survival value was calculated for one patient after appearance of t(14;19)(q32;p13.1) in a relapse specimen.
Results
Clinical Course
The clinical and laboratory data are summarized in Table 1. There were three men and three women, with a median age of 39 years (range, 21–74 years) at the time of diagnosis. Three patients presented to our institution before receiving chemotherapy and three presented with refractory/relapsed disease after undergoing induction chemotherapy at the referring institutions. Upon our institution, all patients underwent a complete physical examination, computed tomography scans, bone marrow aspiration and core biopsy, and lumbar puncture. Laboratory studies included a complete blood count with differential count, coagulation studies, and serum beta-2-microglobulin and lactate dehydrogenase levels.
The patients presented with chief complaints that included fatigue, lower extremity swelling, and bone pain. One patient (Table 1 patient 3) presented with symptoms of hyperleukocytosis and required leukapheresis. At diagnosis, all patients had severe anemia and thrombocytopenia. The median hemoglobin was 8.8 g/dl (range, 4.9–11.3 g/dl; normal range, 14.0–18.0 g/dl) and the median platelet count was 31 k/μl (range, 17–90 k/μl; normal range, 140–440 k/μl). Three patients presented with leukopenia (Table 1; patients 1, 2, and 4), two patients presented with marked leukocytosis (Table 1; patients 3 and 6), and one patient presented with slight leukocytosis (Table 1; patient 5). All patients had circulating blasts. In addition, all patients presented with markedly elevated serum lactate dehydrogenase levels (range, 861–2026 IU/l; median 1378 IU/l; normal range, 313–618 IU/l) and elevated serum beta-2-microglobulin levels (range, 3.0–4.9 mg/l; median 3.9 mg/l; normal range, 0.7–1.8 mg/l). Three patients had mild-to-moderate splenomegaly. No patients had central nervous system involvement or extramedullary disease.
The patients received a variety of therapeutic regimens (Table 2). Four patients (Table 2, patients 1, 3, 4, and 6) received induction therapy with the hyper-CVAD regimen, consisting of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone, alternating with high-dose cytarabine and methotrexate.11 Two of these patients also received rituximab (Table 2; patients 1 and 4). Two patients (Table 2; patients 2 and 5) received induction chemotherapy with an augmented Berlin–Frankfurt–Munster (BFM) regimen, consisting of vincristine, prednisone, l-asparaginase, daunomycin, cytarabine, and methotrexate.12 All patients received central nervous system prophylaxis with alternating doses of methotrexate and cytarabine.11 Patients with relapsed/refractory disease also received treatment with the anti-CD19 antibody, blinatumomab.13
Three patients who were initially diagnosed and treated at our institution (Table 1; patients 1, 2, and 5) all achieved complete remission with induction therapy and two subsequently underwent stem cell transplantation (Table 2). All three patients, however, eventually relapsed. For the two patients who underwent stem cell transplantation (Table 2; patients 1 and 2), one died from sepsis and the other died from graft vs host disease. The third patient (Table 1; patient 5) presented with an initial karyotype of 46,XY,del(12)(p12) and achieved complete remission after chemotherapy. He acquired the t(14;19)(q32;p13.1) as the sole abnormality upon relapse 39 months later. The patient underwent re-induction chemotherapy with augmented BFM and achieved a second complete remission. However, this was followed by a relapse at 2 weeks and the patient subsequently died from sepsis 18 months after the appearance of t(14;19)(q32;p13.1) and 58 months after initial diagnosis.
The three patients who were referred to MD Anderson for refractory disease (Table 1; patients 3, 4, and 6) received induction chemotherapy with hyper-CVAD at the referring institution, and one also received rituximab. One patient achieved a complete response at the referring institution (Table 2; patient 4) and the other two patients had persistent disease (Table 2; patients 3 and 6). These patients were referred to our institution after multiple induction regimens and received treatment at our institution with blinatumomab (anti-CD19). One patient (Table 2; patient 4) achieved a brief complete remission at our institution but relapsed after 2 weeks and died from pneumonia and sepsis. The other patients (Table 2; patients 3 and 6) were unable to achieve complete remission. Patient 3 subsequently died from sepsis and patient 6 is alive with disease.
Overall, four of six patients achieved an initial complete response following induction chemotherapy, but all developed multiple relapses. At the end of the study period, five patients had died from complications of therapy and immunosuppression (infection/sepsis) and one patient was alive with disease. After detection of t(14;19)(q32;p13.1), the median time to relapse was 6 months (range, 2 weeks–12 months) and median survival was 12 months (Table 2).
Morphologic Findings
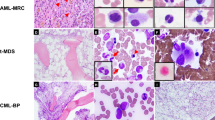
In all cases, the initial diagnostic bone marrow samples were hypercellular (median cellularity 90%; range 80–100%) with sheets of blasts comprising 60–93% of the bone marrow cellularity (median blast percentage 90%; Table 1). The blasts were small- to medium-sized with variably condensed chromatin, prominent nucleoli, and variable amounts of agranular cytoplasm (Figures 1a and b). There was marked trilineage hypoplasia and the residual hematopoietic elements observed were morphologically unremarkable. Bone marrow specimens obtained at relapse showed similar morphologic features.

(Case 2). B acute lymphoblastic leukemia (B-ALL) associated with t(14;19)(q32;p13.1)/IGH-EPOR. (a) Hypercellular bone marrow composed of sheets of blasts (hematoxylin and eosin, × 400). (b). Blasts are of variable size with scant to moderate agranular cytoplasm, irregular nuclear membranes, vesicular chromatin, and prominent nucleoli (Wright–Giemsa, × 500).
Immunophenotypic Findings
Flow cytometry immunophenotypic analysis showed a precursor B-cell immunophenotype in all diagnostic samples. All cases were positive for CD10, CD19, CD20, CD22, CD34, CD38, HLA-DR, and TdT. Additional positive markers assessed included CD9 (3/3), CD13 (3/6), CD33 (2/6), CD58 (4/4), CD66 (4/6) and CD79a (4/6). All cases were negative for CD1a, CD2, surface and cytoplasmic CD3, CD4, CD5, CD7, CD8, CD14, CD15, CD25, CD41, CD56, CD64, CD117, surface immunoglobulin, and myeloperoxidase. Bone marrow specimens obtained at relapse showed a similar, precursor B-cell immunophenotype.
Conventional Cytogenetic and FISH Analyses
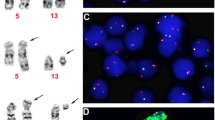
The results of conventional cytogenetic and FISH analyses are summarized in Table 3. Conventional cytogenetic analysis showed a morphologically subtle reciprocal translocation between the long arm of chromosome 14 and the short arm of chromosome 19 (Figure 2). In five out of six patients, conventional cytogenetic analysis demonstrated the t(14;19)(q32;p13.1) in the initial diagnostic specimen. The t(14;19)(q32;p13.1) was detected as the sole cytogenetic abnormality in four patients, and as part of a complex karyotype in two patients. In one patient (Table 3; case 5), cytogenetic analysis of the initial diagnostic specimen revealed 46,XY,del(12)(p12). The t(14;19)(q32;p13.1) was identified as the sole abnormality in a subsequent bone marrow specimen obtained during relapse 3 years after the initial diagnosis. No other recurrent cytogenetic abnormalities were identified in any sample. Conventional cytogenetic analysis showed the t(14;19) in all subsequent specimens with morphologic evidence of disease. All morphologically negative samples and samples with only minimal residual disease detected by flow cytometry showed a normal karyotype without evidence of t(14;19)(q32;p13.1).

(Case 2). B acute lymphoblastic leukemia (B-ALL) associated with t(14;19)(q32;p13.1)/IGH-EPOR. Female karyotype showing abnormal chromosomes (with arrow) der(14)t(14;19) and der(19)t(14;19) resulting from a subtle translocation involving 14q32 and 19p13.1.
FISH analysis using home-brew probes for IGH at 14q32 and EPOR at 19p13.1 showed a single yellow fusion signal on the derivative chromosome 14 (Figure 3). The fusion signal was present in all initial and relapse bone marrow specimens with morphologic evidence of disease and t(14;19)(q32;p13.1) by conventional cytogenetic analysis. FISH analysis was negative in all bone marrow specimens without morphologic evidence of disease and also in specimens with only minimal residual disease detected by flow cytometric immunophenotyping. The percentage of positive nuclei by FISH ranged from 50 to 97% (median 80%). There were no instances of discordance between conventional cytogenetic and FISH results. All patients were Philadelphia chromosome-negative by FISH and/or RT-PCR.

(Case 2). B acute lymphoblastic leukemia (B-ALL) associated with t(14;19)(q32;p13.1)/IGH-EPOR. Fluorescence in situ hybridization (FISH) mapback on a metaphase cell (left) and interphase cells (right) with a t(14;19)(q32;p13.1) using home-brew FISH probes for IGH (green) and EPOR (red). The yellow signal indicates IGH/EPOR rearrangement on a der(14)t(14;19) resulting from a t(14;19)(q32;p13.1).
Discussion
We report the clinicopathologic findings of a series of six cases of B-ALL with t(14;19)(q32;p13.1) and confirmed rearrangement of the IGH and EPOR genes using FISH analysis.
The patients were relatively young adults (median age, 39 years) and all patients presented with high bone marrow cellularity and high blast percentages. All neoplasms had a precursor B-cell immunophenotype, with expression of CD20. Although some authors have linked CD20 expression in B-ALL with higher recurrence rates, recent studies have found no prognostic impact of CD20 expression in Ph-negative B-ALL.14, 15 Five of six (83%) cases showed aberrant expression of myeloid markers, CD13 and/or CD33.
Translocations involving the IGH gene have been identified in 2–3% of B-ALL cases and involve multiple partner genes.3 The functions of the partner genes vary, but most often the partners are lineage-specific transcription factors or involved in proliferative signal transduction.16 Cases of B-ALL with IGH translocations tend to occur in younger patients and have a poor prognosis.3 Translocations involving the IGH locus, including the t(14;19)(q32;p13.1), are often cytogenetically subtle, possibly because of the involvement of the telomeric end of chromosome 14q. Detection of IGH translocations is facilitated by the use of IGH break-apart FISH probes.
The t(14;19)(q32;p13.1) appears to be a stable cytogenetic finding as it was present in every specimen with morphologic evidence of B-ALL, at initial diagnosis and at time of relapse. The translocation was either a sole abnormality or part of a complex karyotype. These results suggest that t(14;19)(q32;p13.1)/IGH-EPOR is a recurrent cytogenetic abnormality in B-ALL, and represents an early, possibly initiating, event.
EPOR encodes for a precursor protein that undergoes post-translational modification to become a 56–57-kDa protein that is eventually transported to the cell surface, making it accessible for binding with erythropoietin. EPOR is normally found on the surface of erythroid progenitors in the bone marrow and interacts with erythropoietin to promote erythropoiesis by activating the JAK2–STAT5 pathway. Transcription of EPOR is normally under the control of the transcription factors GATA-1, Fog1, and SCL/TAL1.7 Ectopic expression of EPOR has been identified in B-ALL with t(12;21)(p13;q22) and ETV6-RUNX1 rearrangement.17, 18 Leukemic blasts in these cases express EPOR and bind erythropoietin, increasing cell survival through activation of the JAK–STAT5 pathway.17 Because of a lack of sufficient genetic material, we were unable to perform testing to confirm the presence of increased EPOR expression in our cohort. However, in their initial report of B-ALL with t(14;19)(q32;p13.1), Russell et al5 demonstrated increased expression of EPOR mRNA in one patient. It is possible that the increased expression of EPOR in B-ALL with t(14;19)(q32;p13.1) confers a cell survival advantage similar to that seen in B-ALL with t(12;21) (p13;q22). It is also possible that t(14;19) confers a survival advantage through a different, as yet undescribed mechanism.
Despite treatment with a variety of aggressive multiagent chemotherapy regimens, including novel anti-CD19 therapy, all patients had a course characterized by short remission duration and multiple relapses. The median overall survival was short, ∼1 year. In the single patient with an initial karyotype that did not include t(14;19), the length of initial complete remission was 39 months. However, after detection of the t(14;19), the patient had a short time to relapse of 2 weeks and died 18 months after detection of the translocation. In the initial description of t(14;19)/IGH-EPOR by Russell et al,5 a 38-year-old patient suffered several relapses, the first of which occurred at 6 months. However, this patient was alive at 13 months. In contrast, a second, 11-year-old patient achieved a sustained complete remission and was alive at 48 months. Our cohort was limited to adult patients. Although the prognosis for adult patients with B-ALL is generally poor, our findings suggest that the t(14;19)(q32;p13.1)/IGH-EPOR confers a worse prognosis. We cannot assess the prognostic effect of t(14;19) in B-ALL of pediatric patients based on the data we present.
Recently, cases of Philadelphia chromosome-negative B-ALL with a unique gene expression profile and an aggressive phenotype similar to Philadelphia chromosome-positive B-ALL have been described. These cases, termed ‘Ph-like ALL’, have been documented mainly in pediatric patients and are associated with gene mutations involved in B-cell development, such as IKZF1, TCF3, EB1, and PAX5.19 Roberts et al6 studied 15 cases of IKZF1-deleted Ph-like B-ALL. All 15 cases had alterations of genes encoding cytokine receptors and regulators of kinase signaling, including one B-ALL with IGH-EPOR rearrangement. On the basis of their findings, Roberts et al 6concluded that the hallmark of Ph-like ALL was the presence of abnormalities in genes encoding regulators of kinase signaling, and that activation of the JAK/STAT5 pathway was a common mechanism of transformation in these cases. Although EPOR lacks direct tyrosine kinase activity, it uses JAK2 as an accessory tyrosine kinase to induce the intracellular signaling cascade, which in turn activates the STAT5, MAPK, and PI3 kinase/AKT signaling pathways.7 Our cohort was limited to adult patients and we lacked sufficient genetic material to perform testing for IKZF1 deletion. However, the decreased overall survival and short time to relapse in our small cohort of patients were similar to those previously reported for IKZF1-deleted, BCR-ABL1-positive B-ALL.20
In summary, t(14;19)(q32;p13.1) involving IGH and EPOR, is a rare, recurrent cytogenetic abnormality in B-ALL that appears to occur more frequently in young adults and has a highly aggressive clinical course. There are no clinical, morphologic, or immunophenotypic characteristics that reliably predict the presence of t(14;19)(q32;p13.1)/IGH-EPOR. The translocation can be subtle and therefore missed by conventional cytogenetic analysis. Use of FISH can facilitate detection of t(14;19) and may allow for better understanding of the true frequency of this aggressive subset of B-ALL.
References
Borowitz MJ, Chan JKC . B lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities In: Swerdlow SH, Campo E, Harris NL et al. (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, 2008, pp 171–175.
Willis TG, Dyer MJ . The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood 2000;96:808–822.
Dyer MJ, Akasaka T, Capasso M et al. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood 2008;115:1490–1499.
Akasaka T, Balasas T, Russell LJ et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2007;109:3451–3461.
Russell LJ, De Castro DG, Griffiths M et al. A novel translocation, t(14;19)(q32;p13), involving IGH@ and the cytokine receptor for erythropoietin. Leukemia 2009;23:614–617.
Roberts KG, Morin RD, Zhang J et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012;22:153–166.
Elliott S, Sinclair AM . The effect of erythropoietin on normal and neoplastic cells. Biologics 2012;6:163–189.
Yin CC, Lin P, Carney DA et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma associated with IgM paraprotein. Am J Clin Pathol 2005;123:594–602.
Jaso J, Thomas DA, Cunningham K et al. Prognostic significance of immunophenotypic and karyotypic features of Philadelphia positive B-lymphoblastic leukemia in the era of tyrosine kinase inhibitors. Cancer 2011;117:4009–4017.
Luthra R, Medeiros LJ . TaqMan reverse transcriptase-polymerase chain reaction coupled with capillary electrophoresis for quantification and identification of bcr-abl transcript type. Methods Mol Biol 2006;335:135–145.
Thomas DA, O'Brien S, Faderl S et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol 2010;8:3880–3889.
Chang JE, Medlin SC, Kahl BS et al. Augmented and standard Berlin-Frankfurt-Munster chemotherapy for treatment of adult acute lymphoblastic leukemia. Leuk Lymphoma 2008;49:2298–2307.
Kantarjian HM . Monoclonal antibodies in adult acute lymphoblastic leukemia. Hematology 2012;17:S52–S54.
Faderl S, O'Brien S, Pui CH et al. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer 2010;116:1165–1176.
Mannelli F, Gianfaldoni G, Intermesoli T et al. CD20 expression has no prognostic role in Philadelphia-negative B-precursor acute lymphoblastic leukemia: new insights from the molecular study of minimal residual disease. Haematologica 2012;97:568–571.
Zhou Y, You MJ, Young KH et al. Advances in the molecular pathobiology of B-lymphoblastic leukemia. Hum Pathol 2012;43:1347–1362.
Torrano V, Proctor J, Cardus P et al. ETV6-RUNX1 promotes survival of early B lineage progenitor cells via a dysregulated erythropoietin receptor. Blood 2011;118:4910–4918.
Inthal A, Krapf G, Beck D et al. Role of the erythropoietin receptor in ETV6/RUNX1-positive acute lymphoblastic leukemia. Clin Cancer Res 2008;14:7196–7204.
Den Boer ML, Van Slegtenhorst M, De Menezes RX et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009;10:125–134.
Martinelli G, Iacobucci I, Storlazzi CT et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol 2009;27:5202–5207.
Acknowledgements
We thank Denise Lovshe, Joanne Cheng, Danielle Saldi and others in the Clinical Cytogenetics Laboratory at MD Anderson Cancer Center for their assistance and support. This study was partially supported by CytoTest.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Jaso, J., Yin, C., Lu, V. et al. B acute lymphoblastic leukemia with t(14;19)(q32;p13.1) involving IGH/EPOR: a clinically aggressive subset of disease. Mod Pathol 27, 382–389 (2014). https://doi.org/10.1038/modpathol.2013.149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.149
Keywords
This article is cited by
-
Childhood B-acute lymphoblastic leukemia: a genetic update
Experimental Hematology & Oncology (2014)
-
ETV6–FLT3 fusion gene-positive, eosinophilia-associated myeloproliferative neoplasm successfully treated with sorafenib and allogeneic stem cell transplant
Leukemia (2014)
