
Abstract
The 2012 USCAP Long Course ‘Malignant Lymphomas—Building on the Past, Moving to the Future’ began with an introduction to lymphoma classification over the last half century and a discussion of our current diagnostic armamentarium, together with a look toward the future. The Rappaport classification, originally published in 1956, was a morphologic classification with few categories. The early 1970s saw a great and tumultuous revolution in the field with the publication of two functional lymphoma classifications that related the malignant lymphomas to the cells of the normal immune system—the Lukes/Collins classification from the United States and the Kiel classification from Professor Lennert and the European Lymphoma Club. With discord abounding, the NCI working formulation, published in 1982, satisfied some but was a step back to a morphologic-based classification. In 1994, the International Lymphoma Study Group published the REAL classification, which reflected state-of-the-art practice for that time, and was shortly followed by preparations for the modern World Health Organization (WHO) classification published in 2001 and revised in 2008. The WHO classification, created by hematopathologists working with the advice and consent of clinical hematologist/oncologists, recognizes numerous distinct entities, defined based on their histopathological, immunophenotypical, molecular/cytogenetic and clinical features. The classification requires use of a multiparameter approach to lymphoma diagnosis although we still rely heavily on histopathology. Immunophenotypical studies, whether using paraffin section immunohistochemistry and/or flow cytometry, are also critical in almost all circumstances. Molecular/cytogenetic techniques that are constantly changing have an increasingly important role, even if not always required. The full impact of next-generation sequencing is yet to be felt but we are beginning to catch a glimpse of what is in our future. Finally, one must not forget the great importance of clinical data in arriving at a diagnosis that best serves the patient, our ultimate goal.
Similar content being viewed by others

Main
The 2012 USCAP Long Course ‘Malignant Lymphomas—Building on the Past, Moving to the Future’ embodied much of what has made the topic of malignant lymphomas one of the great interest to many but also an area that has seen great controversy. As was clearly apparent to those who attended the course, we have moved from a field full of conflict and personal animosity to one with international cooperation and consensus, as reflected in a collegial international faculty trained in a variety of different lymphoma ‘camps’ of the past. This was the fifth long course dealing with lymphoid proliferations, with the first over a half century ago. The title of the 2012 Long Course aimed to underscore how our current understanding of the malignant lymphomas and the practice of diagnostic hematopathology is based on a multilayered foundation built with blood, sweat and even tears by many hematopathologists over the last century but with new layers constantly being added, thanks to new technologies as well as related to new observations based on older methodologies, even including our light microscopes and hematoxylin and eosin-stained slides. It was the aim of this course, and now of this issue of Modern Pathology, to enhance the understanding of where we have come from, where we are now and to provide a hazy glimpse into where we at least think we are going in the future. While exploring some of the very latest scientific discoveries related to the lymphomas, our highest priority was to be certain to provide pragmatic guidance in terms of the practice of state-of-the-art diagnostic hematopathology. We also hoped to stress that pathologists cannot practice in a vacuum, how we must be responsive to the needs of the clinicians and how, working as a team, we can foster the well-being of the patients whom we share.
This introductory manuscript aims to provide a brief review of the long and winding road we have followed on our way to the current World Health Organization (WHO) classification of the malignant lymphomas and a look at our ever-growing diagnostic armamentarium. The manuscripts that follow and that go into much greater detail than this introduction, all represent the individual views of their authors, and have not been officially vetted by those responsible for the WHO classification nor by the Society for Hematopathology or European Association for Haematopathology.
Lymphoma classification over the last half century: a tale of revolution and evolution
It should be acknowledged that as with all other historical reviews, individual perspectives may vary somewhat based on one’s own personal experiences. Furthermore, this review is not intended to be comprehensive, and I apologize to all (at least mostly posthumously) whose classifications are not discussed here. Other much more detailed reviews of the evolution of lymphoma classification have been published over the last few years.1, 2 And while we may all wish for simpler times, I refer readers to ‘A Textbook of Pathology for Students of Medicine’ by JG Adami and J McCrae published almost a century ago,3 who wrote the following about ‘Lymphoma and Lymphomatosis’: ‘It would be unwise to plunge into the vortex of the numerous terms employed to describe the various lymphomatous states, and to lay down what seem to us the right interpretations of these various terms.’
This review begins with the Rappaport classification, initially published in 1956 in a publication on follicular lymphomas (Table 1).4 It was officially published with some changes in Dr Rappaport’s AFIP fascicle in 1966 including the famed but flawed change to histiocytic from reticulum cell for the large-cell lymphomas and the introduction of an undifferentiated category.5 This strictly morphological classification had five and then eight categories and required subsequent distinction of diffuse and nodular (‘follicular’) growth patterns. With the 1970s came a major revolution in this field with the recognition on both sides of the Atlantic that lymphomas are neoplasms of the immune system and should be classified based on the normal cells that they most closely resemble. At that time, this reflected very careful morphological review and some immunophenotypic data. The Lukes/Collins classification was published together with a schema of B- and T-cell differentiation, which was directly related to their proposed classification (Table 2) (Figure 1).6 The Kiel classification, also published in 1974 by K Lennert and the European Lymphoma Club,7 initially divided the lymphomas into cytologically low-grade versus high-grade lymphoid neoplasms, which were then grouped based on the specific cell types involved (Table 3) (Figure 2). Both classifications were subsequently updated8, 9 (as also was the Rappaport classification) although the updated versions will not be presented in this brief overview. The updated Kiel classification divided the lymphomas based first on their B- or T-cell origin. It is important to recognize that, as expressed in the manuscript entitled ‘Concordance of the Kiel and Lukes-Collins classifications of non-Hodgkin’s lymphomas’, Lennert, Collins and Lukes felt that in spite of some specific differences between the two classifications, they were really on the same page.10 In fact, Professor Lennert wrote in an obituary for Dr Lukes that ‘From 1972 on, Dr Lukes and his colleague Dr Robert Collins shared a basic idea with the Kiel group, namely that of deriving malignant lymphomas from the immunologically defined types of lymphocytes. Thus the so-called Lukes-Collins and the Kiel classification evolved simultaneously, the latter introduced by the European Lymphoma Club.’11 Not everyone, however, shared this collegial outlook and there was great consternation over whether the Rappaport classification, one of the functional classifications or one of the other published classifications that existed, should prevail. With classifications proliferating it seemed faster than rabbits, a letter to the Lancet was published with a suggested ‘tongue-in-cheek’ classification of classifications and ultimately the hope for a classification of classifications of classifications.12 One might argue that Charles Dickens had foreshadowed the 1970s hematopathology scene in A Tale of Two Cities when he wrote ‘It was the best of times, it was the worst of times, it was the age of wisdom, it was the age of foolishness….’ A major international study was subsequently undertaken under the auspices of the National Cancer Institute to compare the major existing classifications and provide a way to translate between them, which led to the publication of the NCI Working Formulation in 1982 (Figure 3).13 While some involved in the project believed that the Working Formulation was dead on arrival, others rallied behind it and it became a widely used classification with 10 categories divided into three clinical grades. It was once again a strictly morphologically based classification and included some very heterogeneous categories that included multiple types of lymphomas while dividing up other lymphoma entities into more than one category. The concept seemed to evolve that what was important was just to say if a lymphoma was one of the ‘good’ ones (low grade) or ‘bad’ ones (intermediate or high grade). If only life were that simple!

Robert D Collins at home in ∼1981 (see Figure 3 for picture of RJ Lukes).

Karl Lennert in front of his institute in 1983. (Professor Lennert passed away in August, 2012.)

‘Expert’ and ‘panelist’ pathologists who participated in the NCI Working Formulation Study (from left to right: Koji Nanba, Gerhard Krueger, Ronald Dorfman, Gregory O’Conor, Karl Lennert, Alistair HT Robb-Smith, Henry Rappaport, Martin Sacks, Kristin Henry, Robert J Lukes, Robert J Hartsock and Costan Berard) (photograph contributed by Roger Warnke).
With both of the functional lymphoma classifications aging and without a replacement, an updated set of lymphoma diagnoses borrowing heavily from the prior functional classifications started evolving. For example, the concept of MALT lymphomas first described in the early 1980s was becoming widely accepted and utilized even though not a part of a recognized official lymphoma classification.14 With the need then for an up-to-date biologically meaningful and clinically useful classification and with concerns about a continental divide, the International Lymphoma Study Group was formed and in 1994 published the Revised American-European classification of lymphoid neoplasms (Figure 4).15 The so-called ‘REAL’ classification recognized ‘real entities’ with more than 20 types or provisional types of non-Hodgkin lymphomas plus 4 types and 1 provisional type of Hodgkin lymphoma (Table 4).

International Lymphoma Study Group at meeting in 1992 before publication of their REAL classification (photograph contributed by Roger Warnke).
Soon after the completion of the REAL classification, work began on the modern WHO classification of haematopoieic and lymphoid tumors—an effort supported by the Society for Hematopathology and the European Association for Haematopathology and under the auspices of the WHO and the International Agency for Research on Cancer. The WHO classification was developed by a large number of hematopathologists who obtained the advice and consent of clinical hematologists/oncologists (the so-called Clinical Advisory Committee) including a large meeting of the two groups at Airlie House in Virginia in 1997. The basic philosophy of the WHO classification is to identify distinct clinicopathological entities based on a combination of morphology, immunophenotype, molecular/cytogenetic findings and clinical features. Ultimately, the classification, published in 2001, became ‘the’ classification to use.16 But time marched on. As with all creatures, great and small, the classification aged and the monograph became unavailable. After another major effort, more clinical advisory committee meetings in 2007 (Figure 5) and further editorial meetings extending into 2008, an updated WHO classification and monograph was published in 2008 (Table 5).17 The number of categories of B/T/natural killer (NK)-cell neoplasms recognized almost doubled from 2001 to 2008 and, as also tends to happen with individuals as they age, the monograph became chubbier, adding about 25% more pages. Nevertheless, it was more a matter of the classification continuing to evolve rather than any truly revolutionary new ideas. A detailed discussion of many of the entities included in the classification, together with updates since it was published ∼4 years ago, follow in the other manuscripts in this journal. One must realize, however, that this journal in no way represents a comprehensive review of all the lymphomas that we currently recognize—to do that would have meant revising and publishing a new WHO Bluebook, which is definitely beyond our marching orders. Discussions have now begun on how to handle updating the 2008 edition, without a definite timeline as of this publication.

March 2007 meeting of hematopathologists and Clinical Advisory Committee dealing with revisions for the lymphoid portion of the 2008 World Health Organization classification, Airlie House, Virginia.
The tools of our trade—the hematopathologist’s diagnostic armementarium
Having turned our swords into plowshares with a universally accepted lymphoma classification, we now must turn our attention to using it as best we can (and learning how it can be improved). Given that the classification is based on the concept that we should be diagnosing distinct clinicopathological entities defined by their histopathological, immunophenotypical, molecular/cytogenetic and clinical features, it should not come as a surprise that one needs to use a multiparamenter approach to distinguish lymphomas from reactive disorders and to distinguish the myriad of lymphomas from each other. This, however, does not mean that you need all this information to make a diagnosis in every case! A basic knowledge, therefore, of which ancillary studies are available in 2012, the type of information they yield, their strengths, together with their problems and pitfalls, and when to use which is critical if one is to practice cost-effective high-quality diagnostic hematopathology today. Please note that this review of our current diagnostic armentarium (with an eye to the future) is not intended to be a comprehensive review of all the different methodologies we use and their strengths and weaknesses, but rather a more selective discussion of the most important elements.
Histopathology
Histopathology remains extremely important in 2013 and if you do not know the histopathology/cytology of the lymphomas and lymphoma mimics you will be in deep trouble no matter how many additional ancillary studies you perform. There is growing interest in the use of digital slides so we may be using computer monitors more in the future to view our slides.
Immunophenotypical studies
Immunophenotypical studies are also critical in most cases with two major techniques in use—paraffin section immunohistochemistry (IHC) that, of course, does not require advance tissue preparation and flow cytometric immunophenotypic studies (FCIPS) that are also widely used but do require fresh viable tissue. Whether using IHC and/or FCIPS, it is important to use antibody panels that are neither too small nor too large but just the right size, given the nature of the specimen and situation you are dealing with. This does not mean always using panels that are the same size. Panels that are too small may miss something you did not think about initially or be misleading because of an atypical staining pattern with one of the antibodies utilized. Panels that are too large are costly (to someone), a waste of time and precious resources, and can also be misleading with some stains that should not have been performed leading you down the wrong garden path. Furthermore, they invite more external oversight in our medical decision-making process. A specific suggested basic IHC panel and how it should be interpreted has been discussed elsewhere.18
Paraffin Section Immunohistochemical Studies
Paraffin section immunohistochemical studies have certainly revolutionized the way we practice hematopathology, thanks to the development of so many antibodies for use in formalin-fixed paraffin-embedded tissue sections. These have been developed by many individuals including the late David Mason who realized that if one wanted monoclonal antibodies that worked well in formalin-fixed tissues, one needed to make them with that in mind. We all rely very heavily these days on paraffin section IHC stains that have many strengths, such as the availability of an ever-increasing multitude of paraffin-reactive antibodies including some that are not done using FCIPS and ever-improving detection techniques, the ability to use routinely processed material and the ability to evaluate both the cytologic appearance and growth pattern of positive (and negative) cells, even if sparse or difficult to identify using flow cytometry. Paraffin section IHC also, however, has its problems and pitfalls. Non-plasmacytic κ and λ staining can be accomplished (Figure 6) but is a major challenge and with a moderate number of non-interpretable cases. Many laboratories have stains that reliably only detect cytoplasmic staining, as seen in plasma cells. As there is immunoglobulin in tissues, one must put up with some ‘background’ staining to have sensitive stains. Most antigenic coexpression, especially if on the surface membrane, can only be evaluated when present on major populations. Some antibodies cannot be used on paraffin sections, for example CD103 stains, that are used to help identify hairy cell leukemia and enteropathy-associated T-cell lymphoma. Quantitation is not the easiest, especially if one is looking for small differences in the proportion of positive cells.

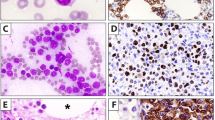
Paraffin section immunohistochemical demonstration of kappa light-chain restriction in a lymph node with interfollicular involvement by chronic lymphocytic leukemia/small lymphocytic lymphoma. (a) The imperfect histological section depicts the same area illustrated in the IHC stains. There is architectural preservation with intact sinuses and a follicle with a germinal center, in addition to an interfollicular expansion of predominantly small lymphocytes. (b) At higher magnification, there are numerous small round lymphocytes with a central ill-defined and somewhat difficult to see proliferation center. (c) There is extensive kappa expression in many areas. (d) At higher magnification, there is kappa positivity both in and adjacent to the germinal center that is on the right. (e) The lambda stain shows most positivity restricted to the residual germinal center and in scattered remaining mantle zone cells, (f) as better seen at higher magnification. (a H&E, original magnification × 10, b H&E, × 40, c–f IHC with hematoxylin counterstain, c and e × 10, d and f × 40).
Paraffin Section In Situ Hybridization (ISH) Stains
Paraffin section ISH stains have a very limited role with their major use being to identify evidence of Epstein–Barr virus infection in tissues using a stain for EBER (EBV-encoded small RNA) and to identify kappa and lambda staining in cells with plasmacytic differentiation when ‘background’ staining makes interpretation of IHC too problematic. The EBER ISH stain is more sensitive than LMP1 immunohistochemical staining for EBV, as LMP1 is only expressed in latent infections with a type II or III latency pattern. In addition, even when present, only a minority of cells may be LMP1 positive. ISH for kappa and lambda mRNA are worth having access to as a backup to IHC stains; however, they are not very sensitive and, in many laboratories, only adequate for assessment of plasmacytic populations.
Flow Cytometric Immunophenotypic Studies
FCIPS are another ancillary technique where there continues to be major changes and progress with the development of more commercially available antibodies, more usable fluorochromes, clinical instruments that can simultaneously analyze the increasing number of fluorochromes and evolution of the all-necessary software (Figure 7). This has led to the provision of more information, the ability to use smaller samples, identification of smaller distinct populations that may be of great clinical importance and very sensitive minimal residual disease testing (not being covered here at all). One can now do a complete basic phenotype using a single tube of stained cells, enhancing, for example, the ability to deal with limited cytology specimens. FCIPS are used extensively in my institution but I also realize that many of the most difficult cases, the kind that get sent off to consultants, are handled without flow cytometric studies (and sometimes misleading flow cytometric studies can actually generate consultations).

Follicular lymphoma analyzed with multicolor flow cytometric studies. A single tube containing seven separately labeled antibodies (for kappa, lambda, CD19, CD20, CD5, CD10 and CD45) demonstrates two populations of B cells among the CD45+ population. One population, representing the lymphoma (CD20+ CD10+ cells highlighted in red), is composed of CD5−, CD10+, lambda light-chain-restricted B cells with variably intense CD19 and somewhat brighter CD20 expression compared with the other polytypic B-cell population, representing the benign non-follicular B cells (CD20+ CD10− cells highlighted in blue), which is CD5− and CD10−. The dimmer CD19 expression on the neoplastic CD10+ B cells is a finding typical of a subset of follicular lymphomas.19 A population of CD5+ T cells (highlighted in black) is also seen. (a) CD19 versus CD20, (b) CD19 versus CD5, (c) CD19 versus CD10, (d) CD10 versus kappa and (e) CD10 versus lambda. (f) A separate tube of cells stained after membrane permeabilization with antibodies to BCL2, CD10 and CD20 demonstrates that the CD10+ B cells (highlighted in red) show stronger BCL2 expression than the other B and T cells present indicating their positivity (BCL2 versus CD10 illustrated).20
FCIPS do have some important major strengths. Multiple phenotypic and two physical (size and ‘complexity’) parameters of individual cells can easily be simultaneously assessed to identify both major and minor normal and abnormal or ‘aberrant’ populations. They remain the best way to identify surface immunoglobulin that is used to identify monotypia and infer monoclonality in many B-cell proliferations. One can detect some antigens not detectable with paraffin section IHC (eg, CD103). Antigen intensity, which has diagnostic/prognostic implications in selected situations, is best evaluated by flow cytometric studies. Identification of ‘aberrant’ T-cell phenotypes used to help diagnose T-cell neoplasms, especially when the neoplastic cells are in sea of normal heterogeneous T cells, is best accomplished with FCIPS. These populations might never be noticed with IHC unless the cells are aggregated or cytologically distinctive. The distinction of T cells with NK cell-associated antigens from true NK cells is often more easily accomplished by flow cytometric studies, again especially if they are not present in great numbers. Whereas, routine FCIPS done without membrane permeabilization detect only surface CD3 that should be lacking on NK cells; both T cells and NK cells will stain for CD3 in paraffin sections with the antibody used by most in clinical practice. In addition, some T cells, like NK cells, are CD5 negative so that pan-T-cell marker is not always useful in making the distinction. CD2 and CD7 are expressed by both T and NK cells, so they are not useful in distinguishing these two subsets. In addition, FCIPS seem to be a more sensitive method to detect CD56 compared with IHC.
FCIPS, however, also have some very major weaknesses. As they are performed on cell suspensions, there is loss of architectural features and the dilutional effect of normal cells sometimes masks the presence of a neoplastic population. Loss of cells of interest owing to fragility and cell death is not infrequent, so one can end up with a very extensive and detailed phenotype of the normal cells present and possibly not even realize there is a neoplasm in their midst (sometimes even if it is a very extensive one). Plasma cells and large lymphoid cells are among the cells most likely to be ‘lost’. Most hematopathologists would agree that FCIPS are not good for picking up Hodgkin lymphoma (recognizing that some would disagree21) or lymphomas without an overtly aberrant phenotype. Some important antigens are not generally evaluated by flow cytometry, eg, cyclin D1. The interpretation of specific findings is not always black and white owing to technical and other factors. Whether an antigen is weakly expressed or not expressed at all is not always clear-cut. Nonspecific staining may masquerade as positivity or obscure other specific staining. Cell ‘doublets’ may be interpreted as populations with an aberrant phenotype (doublets can be excluded as a part of the analysis with the appropriate software). And one must be extremely careful, especially if there is no histopathologic correlate (eg, with fine needle aspirations), as the FCIPS may be very misleading. They may appear to have identified an overt lymphoma with CD10+ or CD5+ light-chain-restricted B cells but actually they represent one of the lesions we now consider to be of uncertain significance (in situ follicular lymphoma22 or mantle cell lymphoma23) or even something benign (florid follicular hyperplasia in a young adult24). Or, they may suggest the wrong type of lymphoma because the phenotype is atypical (eg, a CD10− follicular lymphoma, CD5+ marginal zone lymphoma or CD5− mantle cell lymphoma).
Molecular Studies
Molecular studies are certainly a part of our basic armentarium even if only paraffin-embedded material is available although they are not used nearly as much as histopathology and immunophenotypical studies. The most widely used studies, in terms of lymphoma diagnosis, are those looking for B-cell and T-cell clonality. This is another area where significant progress has been made with the description and availability of BIOMED-2 primers, particularly increasing the yield of finding clones in B-cell neoplasms.25, 26, 27 While they can be extremely useful in selected circumstances, it is important that the results are interpreted properly. This means that, while one does not need to be an expert, one must know enough to appreciate technical and clinical situations that lead to false-positive and false-negative results. Even with molecular studies, the results cannot be considered as absolute for several reasons. Interpretation of whether or not a significant clone is present may be problematic. Monoclonality is not necessarily equivalent to the diagnosis of a malignant lymphoma and lack of a demonstrable clone is not equivalent to assurance that a B- or T-cell proliferation is benign. If a clonal population is present and detected, the molecular studies may still be misleading by suggesting the ‘wrong’ lineage (eg, IGH@ rearrangements in some histiocytic/dendritic neoplasms28, 29, 30, 31) or they may lead to a diagnostic error by identifying a clone that is less important than something else present (eg, finding a restricted T-cell population without a demonstrable immunoglobulin gene rearrangement in a B-cell neoplasm).
Molecular PCR techniques are not generally preferred for assessment of chromosomal translocations at initial diagnosis because of their lower yield compared with cytogenetic fluorescence in situ (FISH) studies (see below).32 An important area to keep one’s eye on is what will undoubtedly be growing interest in using molecular techniques for mutational analyses in the diagnosis and evaluation of lymphoid neoplasms. Mutational analyses have perhaps been more important in the world of myeloid neoplasms up to this point; however, things are changing in that regard. While the ultimate technologies to be utilized remain to be established, as discussed by E Campo in this journal,33 many discoveries are being made, particularly as whole genome/exome sequencing is being explored by many.33, 34, 35, 36, 37 While for some entities, such as chronic lymphocytic leukemia, the mutations being discovered are each present in a relatively small proportion of cases, they may still be of great biological and clinical interest with therapeutic implications.33, 35, 36, 37 Detection of other mutations may be useful for diagnostic purposes, being present in a high proportion of a specific type of lymphoid neoplasm. For example, it has been learned only recently that hairy cell leukemia but very few other B-cell neoplasms have BRAF V600E mutations.38, 39, 40, 41 One must be careful, however. In contrast to some reports of no such mutations in other B-cell lymphoid neoplasms, others do report occasional such cases.42 Just as another example, an L265P mutation in the MYD88 gene has been described as important in the ABC type of DLBCL (29%).43 Of interest diagnostically, it is found in only a small proportion of marginal zone lymphomas and CLL but present in about 90% of lymphoplasmacytic lymphomas, suggesting we may finally have a diagnostic tool with a high yield to help make the latter diagnosis.35, 43, 44 The sequencing technologies can also be used to identify chromosomal translocations.
Cytogenetic Studies
Cytogenetic studies also can be extremely important in the evaluation of lymphoid proliferations. G-banded classical cytogenetic studies are not that widely used because they do require fresh tissue and are labor intensive; however, they can be very informative and you do not have to know specifically what you are looking for (Figure 8a).45 Classical cytogenetic studies are performed in appropriate in-house cases at my institution. Cytogenetic FISH studies using a variety of labeled probes to specific chromosomal regions to detect translocations and numerical abnormalities are used more commonly in part because paraffin-embedded tissue sections or cells extracted from paraffin-embedded material can be utilized.46, 47 They can also be performed on fresh cells (Figure 8b) and air-dried touch imprints. Metaphase FISH can be performed on cultured cell populations. They can also be performed in concert with immunohistochemical stains (so-called FICTION or immunoFISH).47, 48

Follicular lymphoma involving the bone marrow. (a) The classical G-banded cytogenetic karyotype demonstrates a t(14;18)(q32;q21). (b) The interphase FISH study with a dual-color BCL2 break apart probe is used here to confirm the presence of a BCL2 translocation as the same G-banded karyotype would be seen with an IGH@/MALT1 translocation. The cell illustrated demonstrates one normal fusion signal, and one set of split signals with the red-labeled probe that binds 5′ of BCL2 separated from the green-labeled probe that binds 3′ of BCL2. (Contributed by U Surti and M Sherer, Pittsburgh Cytogenetics Laboratory, University of Pittsburgh Medical Center).
And more than just on the horizon in terms of clinical testing are the already well-established high-throughput techniques of array comparative genomic hybridization (aCGH) to detect copy number variations and single-nucleotide polymorphism (SNP) arrays to detect copy number variations and uniparental disomies (Figure 9).49, 50 One may also be able to suggest the possible presence of translocations;51 however, at least currently, this is not the major strength or purpose of these techniques. As indicated above, coming along right behind, and even before aCGH and SNP arrays have been incorporated in many clinical practices, is next-generation sequencing.34, 35 For this manuscript to weigh in on whether this should be included under the umbrella of cytogenetic or molecular testing would be off-topic and, as of 2013, much more dangerous than discussing lymphoma classifications.

SNP analysis reveals copy neutral loss of heterozygosity (uniparental disomy) involving the portion of chromosome 17 that includes the TP53 gene in this Burkitt lymphoma. Although there are two copies of this 17p region, one of the TP53 alleles has been deleted. (Analysis performed by S Branden Van Oss.)
Conclusions
This manuscript has laid the groundwork for the other manuscripts in this journal by presenting an overview of the current WHO classification of lymphomas and how we arrived at it, as well as briefly discussing the tools we need to utilize it, and some of their strengths and weaknesses. The basic message is that we should not feel like things are hopeless, with too much to learn and too many techniques to keep track of, but that, with guidance, we can deal with the advances and celebrate them for what they ultimately will mean for our patients. Specifically, with foundations firmly planted in the hard work done by the forward-thinking hematopathologists of the past, the current WHO classification provides (1) a practical way with agreed upon criteria to do our business in 2013 and agreed upon terminology to communicate with our clinicians and (2) a scaffolding for future evolution of our field together with the expectation that we will never reach our final destination (at least in terms of a lymphoma classification). We also must be realistic and realize that not all cases will neatly fall into one of our innumerable categories. You will need the emotional fortitude to deal with this (and sometimes this is when we all need to get some extra help). Finally, one must recognize and acknowledge that this field is full of change—always has been and always will be. In the words of President John F Kennedy, whose wife died of lymphoma years after his assassination, ‘Change is the law of life. And those who look only to the past or present are certain to miss the future.’
References
Jaffe ES, Harris NL, Stein H et al. Classification of lymphoid neoplasms: the microscope as a tool for disease discovery. Blood 2008;112:4384–4399.
Taylor CR, Hartsock RJ . Classifications of lymphoma; reflections of time and technology. Virchows Arch 2011;458:637–648.
Adami JG, McCrae J . A Textbook of Pathology for Students of Medicine. Lea and Febiger: Philadelphia and New York, 1914.
Rappaport H, Winter WJ, Hicks EB . Follicular lymphoma. A re-evaluation of its position in the scheme of malignant lymphoma, based on a survey of 253 cases. Cancer 1956;9:792–821.
Rappaport H . Tumors of the Hematopoietic System. Armed Forces Institute of Pathology: Washington, D.C., 1966.
Lukes RJ, Collins RD . Immunologic characterization of human malignant lymphomas. Cancer 1974;34 (4 Suppl):1488–1503.
Gerard-Marchant R, Hamlin I, Lennert K et al. Classification of non-Hodgkin’s Lymphomas. Lancet 1974;2:406–408.
Lukes RJ . The immunologic approach to the pathology of malignant lymphomas. Am J Clin Pathol 1979;72 (4 Suppl):657–669.
Stansfeld AG, Diebold J, Noel H et al. Updated Kiel classification for lymphomas [letter] [published erratum appears in Lancet 1988;1(8581):372]. Lancet 1988;1:292–293.
Lennert K, Collins RD, Lukes RJ . Concordance of the Kiel and Lukes-Collins classifications of non-Hodgkin’s lymphomas. Histopathology 1983;7:549–559.
Lennert K . Obituary—Professor Dr. Robert J. Lukes. Ann Hematol 1995;71:103–104.
Kay HE . Letter: classification of non-Hodgkin’s lymphomas. Lancet 1974;2:586.
Anonymous. National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer 1982;49:2112–2135.
Isaacson P, Wright DH . Extranodal malignant lymphoma arising from mucosa-associated lymphoid tissue. Cancer 1984;53:2515–2524.
Harris NL, Jaffe ES, Stein H et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group [see comments]. Blood 1994;84:1361–1392.
Jaffe ES, Harris NL, Stein H, Vardiman JW, (eds) Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, 2001, pp 351.
Swerdlow SH, Campo E, Harris NL et al (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC: Lyon, 2008, pp 439.
Garcia CF, Swerdlow SH . Best practices in contemporary diagnostic immunohistochemistry: panel approach to hematolymphoid proliferations. Arch Pathol Lab Med 2009;133:756–765.
Ray S, Craig FE, Swerdlow SH . Abnormal patterns of antigenic expression in follicular lymphoma: a flow cytometric study. Am J Clin Pathol 2005;124:1–8.
Cook JR, Craig FE, Swerdlow SH . Bcl-2 quantitation by multicolor flow cytometry in neoplastic and benign CD10 positive and CD10 negative B-lymphocytes. Mod Pathol 2002;15:989.
Fromm JR, Thomas A, Wood BL . Flow cytometry can diagnose classical hodgkin lymphoma in lymph nodes with high sensitivity and specificity. Am J Clin Pathol 2009;131:322–332.
Jegalian AG, Eberle FC, Pack SD et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood 2011;118:2976–2984.
Carvajal-Cuenca A, Sua LF, Silva NM et al. In situ mantle cell lymphoma: clinical implications of an incidental finding with indolent clinical behavior. Haematologica 2012;97:270–278.
Kussick SJ, Kalnoski M, Braziel RM et al. Prominent clonal B-cell populations identified by flow cytometry in histologically reactive lymphoid proliferations. Am J Clin Pathol 2004;121:464–472.
Bruggemann M, White H, Gaulard P et al. Powerful strategy for polymerase chain reaction-based clonality assessment in T-cell malignancies Report of the BIOMED-2 Concerted Action BHM4 CT98-3936. Leukemia 2007;21:215–221.
Evans PA, Pott C, Groenen PJ et al. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia 2007;21:207–214.
Langerak AW, Molina TJ, Lavender FL et al. Polymerase chain reaction-based clonality testing in tissue samples with reactive lymphoproliferations: usefulness and pitfalls. A report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2007;21:222–229.
Shao H, Xi L, Raffeld M et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol 2011;24:1421–1432.
Castro EC, Blazquez C, Boyd J et al. Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol 2010;13:225–237.
Chen W, Lau SK, Fong D et al. High frequency of clonal immunoglobulin receptor gene rearrangements in sporadic histiocytic/dendritic cell sarcomas. Am J Surg Pathol 2009;33:863–873.
Feldman AL, Arber DA, Pittaluga S et al. Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008;111:5433–5439.
Espinet B, Bellosillo B, Melero C et al. FISH is better than BIOMED-2 PCR to detect IgH/BCL2 translocation in follicular lymphoma at diagnosis using paraffin-embedded tissue sections. Leuk Res 2008;32:737–742.
Campo E . Whole genome profiling and other high throughput technologies in lymphoid neoplasms—current contributions and future hopes. Mod Pathol 2013, (this issue).
Pasqualucci L, Trifonov V, Fabbri G et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 2011;43:830–837.
Puente XS, Pinyol M, Quesada V et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011;475:101–105.
Wang L, Lawrence MS, Wan Y et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011;365:2497–2506.
Fabbri G, Rasi S, Rossi D et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med 2011;208:1389–1401.
Tiacci E, Trifonov V, Schiavoni G et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–2315.
Tiacci E, Schiavoni G, Forconi F et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood 2012;119:192–195.
Blombery PA, Wong SQ, Hewitt CA et al. Detection of BRAF mutations in patients with hairy cell leukemia and related lymphoproliferative disorders. Haematologica 2012;97:780–783.
Arcaini L, Zibellini S, Boveri E et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood 2012;119:188–191.
Aggarwal N, Swerdlow SH, Kelly LM et al. Thyroid carcinoma-associated genetic mutations also occur in thyroid lymphomas. Mod Pathol 2012;25:1203–1211.
Ngo VN, Young RM, Schmitz R et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011;470:115–119.
Treon SP, Xu L, Yang G et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med 2012;367:826–833.
Cook JR, Shekhter-Levin S, Swerdlow SH . Utility of routine classical cytogenetic studies in the evaluation of suspected lymphomas: results of 279 consecutive lymph node/extranodal tissue biopsies. Am J Clin Pathol 2004;121:826–835.
Cook JR . Paraffin section interphase fluorescence in situ hybridization in the diagnosis and classification of non-hodgkin lymphomas. Diagn Mol Pathol 2004;13:197–206.
Martin-Subero JI, Gesk S, Harder L et al. Interphase cytogenetics of hematological neoplasms under the perspective of the novel WHO classification. Anticancer Res 2003;23:1139–1148.
Sargent RL, Cook JR, Aguilera NI et al. Fluorescence immunophenotypic and interphase cytogenetic characterization of nodal lymphoplasmacytic lymphoma. Am J Surg Pathol 2008;32:1643–1653.
Haraksingh RR, Abyzov A, Gerstein M et al. Genome-wide mapping of copy number variation in humans: comparative analysis of high resolution array platforms. PLoS One 2011;6:e27859.
Krijgsman O, Israeli D, Haan JC et al. CGH arrays compared for DNA isolated from formalin-fixed, paraffin-embedded material. Genes Chromosomes Cancer 2012;51:344–352.
Greisman HA, Hoffman NG, Yi HS . Rapid high-resolution mapping of balanced chromosomal rearrangements on tiling CGH arrays. J Mol Diagn 2011;13:621–633.
Acknowledgements
I would like to thank Dr E Campo for the critical and superb role he played working with me in planning the Long Course and reviewing all of the manuscripts. We would both like to thank all of the speakers who have now contributed a set of excellent manuscripts to this issue of Modern Pathology, and all of the participants who listened to us and asked stimulating questions that hopefully are addressed in these pages. I would also like to thank my principal mentor and friend for over 3 decades, Dr RD Collins, the late Dr AG Stansfeld who introduced me to the Kiel classification as a fellow in the Sherlock Holmes room we shared at St Bartholomew’s Hospital, the late Professor Dr K Lennert and others, whose guidance and help in many ways is responsible for what you see here.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Swerdlow, S. Lymphoma classification and the tools of our trade: an introduction to the 2012 USCAP Long Course. Mod Pathol 26 (Suppl 1), S1–S14 (2013). https://doi.org/10.1038/modpathol.2012.177
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.177
Keywords
This article is cited by
-
Distinct pattern of lymphoid neoplasms characterizations according to the WHO classification (2016) and prevalence of associated Epstein–Barr virus infection in Nigeria population
Infectious Agents and Cancer (2021)
-
Ultrasensitive automated RNA in situ hybridization for kappa and lambda light chain mRNA detects B-cell clonality in tissue biopsies with performance comparable or superior to flow cytometry
Modern Pathology (2018)
-
Lymphoma: current status of clinical and preclinical imaging with radiolabeled antibodies
European Journal of Nuclear Medicine and Molecular Imaging (2017)
-
The influence of rituximab-containing chemotherapy on HCV load in patients with HCV-associated non-Hodgkin’s lymphomas
Annals of Hematology (2017)
