
Abstract
Prostatic carcinoma is a heterogeneous disease with frequent multifocality and variability in morphology. Particularly, prostatic small cell carcinoma is a rare variant with aggressive behavior. Distinction between small cell carcinoma of the prostate and urinary bladder may be challenging, especially in small biopsy specimens without associated prostatic adenocarcinoma or urothelial carcinoma. Recently, gene fusions between ETS genes, particularly ETS-related gene (ERG), and transmembrane protease, serine 2 (TMPRSS2) have been identified as a frequent event in prostate cancer. Thus, molecular methods may be helpful in determining the primary site of small cell carcinoma. Thirty cases of prostatic small cell carcinoma from the authors' archives were studied, among which 13 had concurrent prostatic adenocarcinoma. Tricolor fluorescence in situ hybridization (FISH) was performed on formalin-fixed paraffin-embedded tissue sections with a probe cocktail for 3′/5′ ERG and TMPRSS2. Cases of small cell carcinoma of the bladder and conventional prostatic adenocarcinoma (25 each) were also tested as controls. ERG gene alterations were found only in prostate malignancies and not in benign prostatic tissue or bladder small cell carcinoma. TMPRSS2–ERG gene fusion was found in 47% (14/30) of prostatic small cell carcinoma. Of cases with concurrent prostatic adenocarcinoma, 85% (11/13) had identical findings in both components. In 20% of rearranged cases, the ERG abnormality was associated with 5′ ERG deletion. In 17% (5/30) of cases, gain of the 21q22 locus was present. Two cases showed discordant aberrations in the small cell carcinoma and adenocarcinoma, one with deletion of 5′ ERG and one with gain of chromosome 21q, both in only the adenocarcinoma component. Small cell carcinoma of the prostate demonstrates TMPRSS2–ERG rearrangement with comparable frequency to prostatic adenocarcinoma. In cases with concurrent adenocarcinoma and small cell carcinoma, the majority showed identical abnormalities in both components, indicating a likely common clonal origin. Discordant alterations were present in rare cases, suggesting that acquisition of additional genetic changes in multifocal tumors may be responsible for disease progression to a more aggressive phenotype. TMPRSS2–ERG fusion is absent in bladder small cell carcinoma, supporting the utility of FISH in distinguishing prostate from bladder primary tumors and identifying metastatic small cell carcinoma of unknown origin.
Similar content being viewed by others


Main
Prostatic carcinoma is a heterogeneous disease process with a wide spectrum of light microscopic morphologic features, biological behavior,1, 2 and frequent multifocality.3 In particular, small cell carcinoma of the prostate is a rare and aggressive variant, comprising ∼0.5–2.0% of prostatic carcinoma,2, 4, 5, 6 that exhibits unique clinicopathological characteristics, including frequent metastasis to visceral and other sites uncommon for prostate cancer, such as liver, lung, and brain. Patients typically are found to have lower levels of serum prostate-specific antigen and a poorer response to androgen deprivation therapy.2, 7, 8, 9, 10, 11 Because of its rarity and unusual behavior, several differential diagnostic dilemmas may accompany the diagnosis of small cell carcinoma of the prostate, particularly identification of a small cell carcinoma tumor originating from an unknown primary site10 and distinguishing prostatic primary tumors from those of the urinary bladder. Recent studies have identified gene fusions between members of the ETS family of genes and transmembrane protease, serine 2 (TMPRSS2) to be a significant event in prostate cancer;12, 13, 14, 15, 16, 17, 18, 19 in particular, fusion of transcriptional regulator ETS-related gene (ERG) and TMPRSS2. Interestingly, these fusions appear to represent a specific early event in prostatic carcinogenesis15, 20, 21 seen in many of the variant histological forms of prostatic carcinoma22, 23, 24, 25 but absent in epithelial neoplasms of other organs.14 Therefore, molecular testing for ERG–TMPRSS2 rearrangement may be helpful for not only resolving the organ of origin in an individual case, but also more globally, elucidating the histogenesis of prostatic small cell carcinoma.
Materials and methods
Thirty cases of prostatic small cell carcinoma were retrieved from the archives of the authors' institutions, including tumors with and without concurrent prostatic acinar adenocarcinoma for evaluation of ERG–TMPRSS gene rearrangement by fluorescence in situ hybridization (FISH). Cases without associated acinar adenocarcinoma were included on the basis of integration of clinical data and ancillary testing, including serum prostate-specific antigen level, immunohistochemical findings, and clinicopathological absence of small cell carcinoma of another organ. In addition, 25 cases of urinary bladder small cell carcinoma (published in previous studies) and 25 cases of prostatic adenocarcinoma without a small cell component were tested comparatively as controls. Four micrometer-thick sections were obtained from formalin-fixed paraffin-embedded specimen blocks representative of the tumors for use in light microscopic evaluation and FISH studies.
Slides were deparaffinized with two 15-min washes in xylene and subsequently washed twice with 100% ethanol for 10 min each and air dried. The sections were heated at 95°C in 0.1 mM citric acid (pH 6) solution (Invitrogen, Carlsbad, CA) for 10 min, rinsed with distilled water for 3 min, and washed with 2 × saline-sodium citrate (SSC) for 5 min. Tissue digestion was performed by applying 0.4 ml of pepsin (Sigma, St Louis, MO, USA) solution (4 mg/ml in 0.9% NaCI in 0.01 N HC1) to each slide and incubating the slides in a humidified box for 40 min at 37°C. The slides were rinsed with distilled water for 5 min, washed with 2 × SSC for 5 min, and then air dried. A probe cocktail including BAC clones RP11-476D17-gold (3′ ERG), RP11-95121-green (5′ ERG) and RP11-35C4-aqua (TMPRSS2) (Empire Genomics, Buffalo, NY, USA) diluted 1:25 in tDenHyb2 (Insitus, Albuquerque, NM, USA) was prepared.
Five microliters of diluted probe were applied to each slide; coverslips were placed over the slides and sealed with rubber cement. The slides were denatured at 80°C for 10 min and hybridized at 37°C overnight. The coverslips were removed and the slides were extensively washed with two 0.1 × SSC/1.5 M urea solutions at 45°C for 20 min, in 2 × SSC at 45°C for 10 min, and then in 2 × SSC/0.1% NP40 at 45°C for 10 min. Finally, the slides were washed with 2 × SSC at room temperature for 5 min, air dried, counterstained with 10 μl DAPI/Antifade (DAPI in Fluorguard, 0.5 g/ml, Insitus) and sealed with nail polish.
The hybridized slides were observed and documented using a MetaSystem system (Belmont, MA, USA) under × 100 oil objective. The images were acquired with a CCD camera and analyzed with Metasystem Isis software (Belmont). The following filters were used: SP-100 for DAPI, FITC MF-101 for spectrum green, Gold 31003 for spectrum gold, and Aqua 31036V2 for spectrum aqua signals. Signals from each color channel (probe) were counted under false color, with computerized translation of each color channel into blue, green, red, or aqua. Five sequential focus stacks with 0.3 μm intervals were acquired and integrated into a single image to reduce thickness-related artifacts. Evaluation and analysis of the cases were carried out by two pathologists separately (LC and SZ) (Figure 1). A minimum of 100 nonoverlapping cancer cells was evaluated for each case, totalling 100–200 cells per case. Only if >90% of cells demonstrated sufficient signal was the slide considered to be qualified for counting. Cases with ERG signal abnormalities in ≥20% of the tumor cell population were considered to be positive, based upon reported ERG abnormalities in normal epithelium of prostatic glands, varying thresholds between 10 and 50%, experience with other FISH probes in our laboratory, and taking into consideration the potential truncation artifact at ∼20% of cells.

Schematic representation of ERG rearrangement identification by tricolor FISH. The red probe hybridizes to the 3′ sequence of ERG, the green probe hybridizes to the 5′ sequence of ERG, and the aqua probe hybridizes to TMPRSS2. (a) Wild-type (normal) findings at the 21q22 locus show a triplet of overlapping red and green signals with aqua signals spaced at a variable distance from the red–green pair. (b) Cells with ERG–TMPRSS2 rearrangement exhibit splitting (separation) of the red–green signal pair, accompanied by a fused red–aqua signal for one allele. (c) A subset of cases with rearrangement showed loss of the corresponding green signal for one allele, indicating 5′ ERG deletion.
Results
Thirty cases of small cell prostatic carcinoma were retrieved (Table 1), including 13 cases (43%) with a concurrent component of prostatic adenocarcinoma. FISH revealed three major patterns at the 21q22 locus: (1) the wild-type pattern demonstrated two sets of triplet green (ERG 5′), red (ERG 3′), and aqua (TMPRSS2) signals in each cell; (2) in ERG–TMPRSS2 gene fusion, one allele showed a red–aqua signal doublet and a separate green signal, while the other 21q22 locus exhibited preservation of the wild-type pattern; and (3) a subset of cases with ERG–TMPRSS2 gene fusion showed a red–aqua signal doublet with loss of the corresponding green signal, and the other allele showed a wild-type 21q22 locus. (Figure 1) A minority of cases (17%), with and without ERG rearrangement, showed copy number increase of the 21q22 locus.
ERG gene alterations were found only in prostate malignancies and not observed in benign prostate tissue. Of the 25 cases of urinary bladder small cell carcinoma examined, none showed ERG abnormalities. TMPRSS2–ERG gene fusion was found in 47% (14/30) of prostatic small cell carcinoma. Of the 13 cases that included a component of concurrent prostatic adenocarcinoma, 85% (11/13) had identical findings in both the small cell and adenocarcinoma components. (Figure 2) In 10% (3/30) of total cases, deletion of 5′ ERG was detected, comprising 20% (3/15) of the cases with ERG abnormalities in either component. In 17% of cases (5/30), gain of the 21q22 locus was present, comprising 20% (3/15) of cases with ERG abnormalities in either component. Two cases showed gain of 21q22 without ERG rearrangement and two cases showed discordant aberrations in the small cell carcinoma and adenocarcinoma. Of these two discordant cases, one (case 17) showed TMPRSS2–ERG fusion in both components, accompanied by gain of the 21q22 locus; however, deletion of 5′ ERG was present in only the adenocarcinoma component. In the other discordant case (case 16), TMPRSS2–ERG fusion was present only in the adenocarcinoma component and associated with 21q22 gain. In the 25 control cases of conventional prostatic adenocarcinoma without a small cell component, 50% showed gene fusion, three with loss of 5′ ERG.


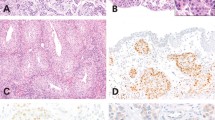
Morphology and ERG gene rearrangement by FISH in small cell carcinoma and adenocarcinoma of the prostate. (a) Low magnification shows admixed small cell carcinoma and concurrent prostatic acinar adenocarcinoma. (b) The wild-type (normal) pattern of ERG demonstrates proximate or fused ERG 3′ (red) and ERG 5′ (green) signals with TMPRSS2 (aqua) either adjacent to or slightly separated from the red–green signal pair. (c) The small cell carcinoma cells are arranged in cords, nests, or sheets, with the nucleus showing prominent hyperchromasia, nuclear molding, small punctate nucleoli, and brisk mitotic activity. (e) In contrast, the typical prostatic adenocarcinoma component retains the characteristic small, round glands. ERG gene rearrangements were detectable by FISH in both small cell (d) and adenocarcinoma components (f). In the typical rearrangement, the green signal (ERG 5′, thin arrows) is separated from the red signal with red and aqua signals approximated (ERG 3′–TMPRSS2 fusion, thick arrows (d, f)). In other cases, rearrangement was associated with a red–aqua signal doublet, with loss of the corresponding green signal (g). A subset of cases with and without rearrangement demonstrated copy number gain at the 21q22 locus (h).
Discussion
Carcinogenesis in the prostate is a complex and heterogeneous disease process exhibiting a wide spectrum of light microscopic morphological features and biological behavior.1, 2 In contrast to tumors of many other organs, multifocal prostate cancer is the rule rather than the exception.3, 26 Small cell carcinoma in particular is a unique variant with behavior distinct from that of typical prostatic adenocarcinoma. Its light microscopic appearance is indistinguishable from small cell carcinoma of other organs, such as the lung; however, its histogenesis remains poorly understood.2, 27, 28, 29 To gain insight into its development and also to evaluate the diagnostic utility of molecular testing in this setting, we studied rearrangement of the ERG and TMPRSS2 genes at the 21q22 locus by FISH in a series of 30 cases of prostatic small cell carcinoma.
The histogenesis of prostatic small cell carcinoma has been long debated. Cells with neuroendocrine differentiation are demonstrable in many or all prostatic carcinomas, as well as in normal prostatic glands, although their relationship to the tumor cells in small cell carcinoma has been incompletely elucidated.2, 27, 28 Some authors have postulated an origin of the tumor directly from stem cells, based on the frequent absence of immunohistochemical staining with organ-specific markers of differentiation, such as prostate-specific antigen, coupled with the exceedingly high proliferation rate, greater than that of dedifferentiated adenocarcinoma.29, 30 In addition, the relationship between poorly differentiated adenocarcinoma with ‘small cell’ morphology and true neuroendocrine small cell carcinoma has been debated.30 Although it is true that many cases are negative for prostate-specific markers, a significant subset of cases show positivity for prostate-specific antigen (17–19%), prostein (P501S, 28%), or prostate-specific membrane antigen (25%).10, 11 This finding, combined with the coexistence of an acinar adenocarcinoma component in another distinct subset of cases27 and identical TP53 mutations in both components,31 suggests that small cell carcinoma may originate from dedifferentiation of conventional adenocarcinoma.
To more thoroughly evaluate this question, we compared TMPRSS2–ERG fusion status in cases of concurrent prostatic acinar adenocarcinoma and small cell carcinoma. We found that prostatic small cell carcinoma showed ERG rearrangement with comparable frequency to prostate cancer in general16, 32, 33 (47%). We found identical abnormalities in adenocarcinoma and small cell carcinoma for 85% of cases; however, a small subset of cases showed discordance of FISH abnormalities in the two components. In one case (case 17), both small cell and adenocarcinoma components showed gene fusion with 21q22 gain; however, only the adenocarcinoma component showed deletion of 5′ ERG. In the second case, (case 16) gene fusion was detected only in the acinar adenocarcinoma component, associated with 21q22 gain. Along these lines, Barry et al 26 found that the majority of cases of multifocal prostate cancer showed homogeneity of TMPRSS2–ERG fusion status within a single tumor focus, while comparing separate tumor foci yielded differences in the molecular abnormalities. Other authors have noted similar findings.34, 35 Our results support the hypothesis that in most cases, acinar adenocarcinoma and small cell carcinoma arise from a common clonal origin through dedifferentiation. The presence of occasional discordance in the two tumor components is in keeping with the tendency of the prostate gland to harbor multiple spatially separate and clonally distinct tumor foci.3 As such, the two morphologically distinct elements may have arisen separately from their own precursor lesions.
In 20% of cases with rearrangement, we found gene fusion to be associated with deletion of 5′ ERG, a somewhat lower rate than has been reported in other studies including cases of prostatic small cell carcinoma.22, 24 However, this finding has been examined in only a relatively small number of cases thus far. This discrepancy may be attributable to a variety of factors, such as varying patient population and methodology/threshold setting, as well as sample size. Using the FISH method, a significant spectrum of probe sets has been utilized, complicating accurate comparison.23, 36, 37 Further investigation in this area may reveal differing mechanisms of gene fusion that occur preferentially in small cell carcinoma as compared with typical acinar adenocarcinoma.
Other avenues of utility for FISH studies include the differentiation of metastatic small cell carcinoma originating from an unknown primary site. In some cases, definitively establishing an origin of small cell carcinoma from the prostate gland may be very challenging. As noted above, usual immunohistochemical markers of prostatic differentiation may be negative in prostatic small cell carcinoma.10, 11 Further compounding this problem, thyroid transcription factor-1 is positive in a significant number of prostatic small cell carcinomas,10, 11 limiting the utility of the marker in distinguishing them from pulmonary small cell carcinomas. In such circumstances, our findings support the use of FISH for TMPRSS2–ERG gene fusion as a specific marker of prostatic origin.
Furthermore, urinary bladder small cell carcinoma may also represent a source of diagnostic difficulty. Tumors originating in the prostate can be difficult to discern pathologically from those originating in the bladder and involving the prostate, and vice versa, especially if a component of differentiated urothelial or prostatic carcinoma is absent. As some authors have noted differences in behavior between tumors of the two primary sites,38 this distinction may be in some cases clinically or prognostically useful. We found absence of TMPRSS2–ERG fusion in all of the 25 studied cases of urinary bladder small cell carcinoma, supporting the utility of FISH in resolving this differential diagnosis.
In 17% of cases, we noted copy number gain at the 21q22 locus, comprising 20% of cases with rearrangement in either component. The significance of this finding is not completely understood. Some authors have noted copy number gain associated with 5′ ERG deletion to correlate with more aggressive disease;39, 40 however, others have noted that copy number gains are associated with generalized chromosomal instability and a non-diploid status at other chromosomal loci.41 Therefore, the unfavorable behavior of these tumors may be a function of non-diploid/aneuploid status. In support of the latter hypothesis, two of our cases of small cell carcinoma showed copy number increase without other ERG rearrangement, in addition to the three other cases (five total) in our study that showed copy number gain, one with 5′ ERG deletion. If indeed the combination of 5′ ERG deletion and copy number gain is associated with more aggressive behavior in prostate cancer, our results suggest that the transformation to small cell carcinoma need not necessarily progress through this pathway. Copy number increase alone or in combination with other ERG abnormalities may be an indicator of increased chromosomal instability and may represent one of the pathways to development of an aggressive tumor.
In summary, prostatic small cell carcinoma is an interesting and aggressive neoplasm associated with a poor prognosis. Its histogenesis has long been debated. Our findings are in keeping with the hypothesis that prostatic small cell carcinoma arises from dedifferentiation of typical acinar adenocarcinoma, although a small subset of cases show variation between tumor components in their genetic abnormalities, perhaps due to the multifocal carcinogenesis typical of prostate cancer. Differentiation of prostatic from urinary bladder small cell carcinoma in some cases may be achieved based on the presence of TMPRSS2–ERG gene fusion, although not all prostate cancers are positive for this abnormality. Similarly, determination of prostatic origin in metastatic small cell carcinoma may be achieved in many cases using this method.
References
Epstein JI, Algaba F, Allsbrook WC, et al. Acinar adenocarcinoma. In: Eble JN, World Health Organization, International Agency for Research on Cancer (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press: Lyon, 2004, pp 162–192.
di Sant'Agnese PA, Egevad L, Epstein JI, et al. Neuroendocrine tumors. In: Eble JN, World Health Organization, International Agency for Research on Cancer (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press: Lyon, 2004, pp 207–208.
Andreoiu M, Cheng L . Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum Pathol 2010;41:781–793.
Helpap B, Kollermann J, Oehler U . Neuroendocrine differentiation in prostatic carcinomas: histogenesis, biology, clinical relevance, and future therapeutical perspectives. Urol Int 1999;62:133–138.
Stein ME, Bernstein Z, Abacioglu U, et al. Small cell (neuroendocrine) carcinoma of the prostate: etiology, diagnosis, prognosis, and therapeutic implications--a retrospective study of 30 patients from the rare cancer network. Am J Med Sci 2008;336:478–488.
Palmgren JS, Karavadia SS, Wakefield MR . Unusual and underappreciated: small cell carcinoma of the prostate. Semin Oncol 2007;34:22–29.
Wenk RE, Bhagavan BS, Levy R, et al. Ectopic ACTH, prostatic oat cell carcinoma, and marked hypernatremia. Cancer 1977;40:773–778.
Amato RJ, Logothetis CJ, Hallinan R, et al. Chemotherapy for small cell carcinoma of prostatic origin. J Urol 1992;147:935–937.
Spiess PE, Pettaway CA, Vakar-Lopez F, et al. Treatment outcomes of small cell carcinoma of the prostate: a single-center study. Cancer 2007;110:1729–1737.
Wang W, Epstein JI . Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol 2008;32:65–71.
Yao JL, Madeb R, Bourne P, et al. Small cell carcinoma of the prostate: an immunohistochemical study. Am J Surg Pathol 2006;30:705–712.
Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644–648.
Tomlins SA, Mehra R, Rhodes DR, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res 2006;66:3396–3400.
Scheble VJ, Braun M, Beroukhim R, et al. ERG rearrangement is specific to prostate cancer and does not occur in any other common tumor. Mod Pathol 2010;23:1061–1067.
Cerveira N, Ribeiro FR, Peixoto A, et al. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia 2006;8:826–832.
Perner S, Demichelis F, Beroukhim R, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res 2006;66:8337–8341.
Albadine R, Latour M, Toubaji A, et al. TMPRSS2-ERG gene fusion status in minute (minimal) prostatic adenocarcinoma. Mod Pathol 2009;22:1415–1422.
Carver BS, Tran J, Gopalan A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 2009;41:619–624.
Iljin K, Wolf M, Edgren H, et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res 2006;66:10242–10246.
Clark J, Attard G, Jhavar S, et al. Complex patterns of ETS gene alteration arise during cancer development in the human prostate. Oncogene 2008;27:1993–2003.
Mosquera JM, Perner S, Genega EM, et al. Characterization of TMPRSS2-ERG fusion high-grade prostatic intraepithelial neoplasia and potential clinical implications. Clin Cancer Res 2008;14:3380–3385.
Han B, Mehra R, Suleman K, et al. Characterization of ETS gene aberrations in select histologic variants of prostate carcinoma. Mod Pathol 2009;22:1176–1185.
Scheble VJ, Braun M, Wilbertz T, et al. ERG rearrangement in small cell prostatic and lung cancer. Histopathology 2010;56:937–943.
Guo CC, Dancer JY, Wang Y, et al. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum Pathol 2011;42:11–17.
Falzarano SM, Navas M, Simmerman K, et al. ERG rearrangement is present in a subset of transition zone prostatic tumors. Mod Pathol 2010;23:1499–1506.
Barry M, Perner S, Demichelis F, et al. TMPRSS2-ERG fusion heterogeneity in multifocal prostate cancer: clinical and biologic implications. Urology 2007;70:630–633.
Schron DS, Gipson T, Mendelsohn G . The histogenesis of small cell carcinoma of the prostate. An immunohistochemical study. Cancer 1984;53:2478–2480.
Abrahamsson PA . Neuroendocrine differentiation in prostatic carcinoma. Prostate 1999;39:135–148.
Simon RA, di Sant'Agnese PA, Huang LS, et al. CD44 expression is a feature of prostatic small cell carcinoma and distinguishes it from its mimickers. Hum Pathol 2009;40:252–258.
Helpap B, Kollermann J . Undifferentiated carcinoma of the prostate with small cell features: immunohistochemical subtyping and reflections on histogenesis. Virchows Arch 1999;434:385–391.
Hansel DE, Nakayama M, Luo J, et al. Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate 2009;69:603–609.
Esgueva R, Perner S, C JL, et al. Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in a large prostatectomy cohort. Mod Pathol 2010;23:539–546.
Mosquera JM, Mehra R, Regan MM, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res 2009;15:4706–4711.
Furusato B, Gao CL, Ravindranath L, et al. Mapping of TMPRSS2-ERG fusions in the context of multi-focal prostate cancer. Mod Pathol 2008;21:67–75.
Mehra R, Han B, Tomlins SA, et al. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer Res 2007;67:7991–7995.
Fine SW, Gopalan A, Leversha MA, et al. TMPRSS2-ERG gene fusion is associated with low Gleason scores and not with high-grade morphological features. Mod Pathol 2010;23:1325–1333.
Zhang S, Pavlovitz B, Tull J, et al. Detection of TMPRSS2 gene deletions and translocations in carcinoma, intraepithelial neoplasia, and normal epithelium of the prostate by direct fluorescence in situ hybridization. Diagn Mol Pathol 2010;19:151–156.
Mackey JR, Au HJ, Hugh J, et al. Genitourinary small cell carcinoma: determination of clinical and therapeutic factors associated with survival. J Urol 1998;159:1624–1629.
Attard G, Clark J, Ambroisine L, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008;27:253–263.
Mehra R, Tomlins SA, Yu J, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res 2008;68:3584–3590.
Gopalan A, Leversha MA, Satagopan JM, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 2009;69:1400–1406.
Acknowledgements
Dr Jiaoti Huang is supported by UCLA SPORE in Prostate Cancer, Prostate Cancer Foundation Challenge Award and Creativity Award, and Department of Defense Prostate Cancer Research Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Williamson, S., Zhang, S., Yao, J. et al. ERG–TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: evidence supporting monoclonal origin. Mod Pathol 24, 1120–1127 (2011). https://doi.org/10.1038/modpathol.2011.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.56
Keywords
This article is cited by
-
Small cell carcinoma of the prostate after low-dose-rate brachytherapy: a case report
Journal of Medical Case Reports (2020)
-
EGFR-upregulated LIFR promotes SUCLG2-dependent castration resistance and neuroendocrine differentiation of prostate cancer
Oncogene (2020)
-
Integrated immunohistochemical and molecular analysis improves diagnosis of high-grade carcinoma in the urinary bladder of patients with prior radiation therapy for prostate cancer
Modern Pathology (2020)
-
Lineage relationship between prostate adenocarcinoma and small cell carcinoma
BMC Cancer (2019)
-
Neuroendocrine tumors of the prostate
Modern Pathology (2018)
