
Abstract
The majority of lung adenocarcinoma patients with epidermal growth factor receptor- (EGFR) mutated or EML4–ALK rearrangement-positive tumors are sensitive to tyrosine kinase inhibitors. Both primary and acquired resistance in a significant number of those patients to these therapies remains a major clinical problem. The specific molecular mechanisms associated with tyrosine kinase inhibitor resistance are not fully understood. Clinicopathological observations suggest that molecular alterations involving so-called ‘driver mutations’ could be used as markers that aid in the selection of patients most likely to benefit from targeted therapies. In this review, we summarize recent developments involving the specific molecular mechanisms and markers that have been associated with primary and acquired resistance to EGFR-targeted therapy in lung adenocarcinomas. Understanding these mechanisms may provide new treatment avenues and improve current treatment algorithms.
Similar content being viewed by others

Main
Approximately 85–90% of all cases of lung cancer are carcinomas of non-small cell type.1, 2, 3 These tumors can be further classified into several major histological subtypes, including adenocarcinoma, squamous cell carcinoma, large cell carcinoma, adenosquamous cell carcinoma, and sarcomatoid carcinoma.4 In recent years, attention has been paid to the role that ‘driver mutations,’ such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK), have in the tumorigenesis of adenocarcinomas, and their potential use as targets for therapy.5, 6, 7, 8, 9 Recent data suggest EGFR may also serve as a prognostic factor, in addition to its role as a predictive factor, as patients-bearing EGFR mutations have shown favorable clinical outcomes even with conventional chemotherapy.10, 11, 12, 13
EGFR and members of its family have an important role in carcinogenesis through their involvement in the modulation of cell proliferation, apoptosis, cell motility, and neovascularization.12, 13, 14, 15, 16 EGFR alterations have been implicated in the pathogenesis and progression of many malignancies.13, 17, 18, 19, 20, 21 The incidence of EGFR mutations in unselected tumors with non-small cell histology ranges from 10 to 50%, depending upon the ethnic makeup of the patient population and the detection methods used for mutation analysis; 95% of such mutations have been found in adenocarcinomas.12, 13, 16, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 Although the exact molecular mechanisms resulting from these somatic mutations are not completely understood, it seems clear that mutant EGFR has enhanced tyrosine kinase activity. Tyrosine kinase is an enzyme that transports phosphates from adenosine triphosphate (ATP) to a protein's tyrosine residue. Although these cases are most often attributed to EGFR mutations, they may also result from increased gene copy number or increased EGFR protein expression.35, 36 EGFR tyrosine kinase inhibitors (TKIs) competitively block the binding of ATP to the catalytic site in the tyrosine kinase domain of EGFR, subsequently inhibiting autophosphorylation. The process blocks downstream signaling and results in dramatic antitumor activity for a subset of lung adenocarcinoma patients.
EGFR alterations have prompted the development of two classes of anti-EGFR agents: monoclonal anti-EGFR antibodies (such as cetuximab, panitumumab, etc) and small molecule TKIs directed against EGFR tyrosine kinase (such as gefitinib, erlotinib, etc). Clinical trials were initiated that employed novel agents targeting EGFR tyrosine kinase. The results of the clinical trials indicated that many of the tumors harboring mutant EGFR are highly sensitive to EGFR TKIs, with up to 70% demonstrating a significant clinical response.5, 28, 29, 37, 38, 39 Recent studies have provided more compelling evidence of the clinical benefits of anti-EGFR treatment in the appropriate setting.13, 15, 22, 38, 40, 41, 42, 43, 44, 45, 46, 47, 48 Evidence from the large phase III randomized Iressa Pan-Asia Study trial and other phase III trials have prompted the American Society of Clinical Oncology to issue a provisional clinical opinion recommending the testing of EGFR mutational status in patients being considered for first line EGFR TKI therapy owing to their demonstrated benefit on progression-free survival.22, 41 Of note, they caution that no definitive benefit has been shown in patients treated with first-line TKIs in regards to overall survival.22
Biomarkers to predict which patients might benefit from targeted therapy are urgently needed. Pathologists have a central role in the process of determining appropriate testing of these tumors and in the interpretation of the test results. In this review, we summarize the most recent developments involving the specific molecular mechanisms and markers that have been associated with primary and acquired resistance to EGFR-targeted therapy, which may lead to new, more effective treatment possibilities and may augment the currently used treatment algorithms.
EGFR alterations in lung cancer
EGFR Mutations
EGFR is located at chromosome 7p11.2, spans about 200 kb, and contains 28 exons. The gene encodes a protein of 464 amino acids.49, 50 EGFR is composed of an N-terminal extracellular ligand-binding domain, a transmembrane lipophilic segment, and a C-terminal intracellular region containing a tyrosine kinase domain. The EGFR tyrosine kinase modulates cell proliferation and survival through auto-activation of EGFR itself, or through two downstream intermediate pathways: the PIK3CA/AKT1/MTOR pathway and the RAS/RAF1/MAP2K1/MAPK1 pathway.51 Upon ligand binding to EGFR, the receptors form homodimers or heterodimers, which activate their intrinsic intracellular protein-tyrosine kinase. The ligand binding-induced dimerization results in cross-autophosphorylation of key tyrosine residues in the cytoplasmic domains, which function as docking sites for downstream signal transducers.36 This activation of EGFR initiates signaling cascades involving several downstream pathways, which induce crucial cellular responses, such as proliferation, differentiation, motility, and survival13, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 (Figure 1).

The EGFR-signaling pathway. EGFR is composed of an extracellular domain, a transmembrane lipophilic segment, and an intracellular region containing tyrosine kinase domains that occupy exons 18–24. The binding of ligands to EGFR results in autophosphorylation of key tyrosine residues in the tyrosine kinase domain and activates tyrosine kinase activity, which further activates the downstream PIK3CA/AKT1/MTOR and RAS/RAF1/MAP2K1 pathways. The aberrant signaling influences several key aspects including cell proliferation, apoptosis, migration, survival, and more complex processes such as angiogenesis.
EGFR mutations, which are associated with objective responses to single-agent TKI therapy in lung adenocarcinomas, are preferentially observed in a specific subset of patients: females of East Asian ethnicity who have never smoked and who have adenocarcinoma with lepidic growth pattern (formerly bronchioloalveolar carcinoma).5, 6, 14, 63, 64
In adenocarcinomas, the majority of mutations have been identified in exons 18–21 of the EGFR gene.9, 65, 66 These mutations can be roughly classified into three major categories: in-frame deletions in exon 19, insertion mutations in exon 20, and missense mutations in exons 18–21 (Figure 2). Different EGFR mutations have different signaling properties, but most mutations affect the ATP-binding cleft, where targeting TKIs compete for binding.58 The most frequent mutations were located at exon 19 and exon 21. There are over 20 variant types of exon 19 deletions, with the most common including delE746-A750, delL747-T751insS, and delL747-P753insS. L858R, in exon 21, is the second most frequent mutation. Additional mutations are located at exon 18 including G719C, G719S, G719A, and S720F and mutations found in exon 21 including L861Q and L861R. The exon 20 insertions frequently associated with EGFR-TKI non-responsiveness, including D770-N771insNPG, D770-N771insSVQ, D770-N771insG, and point mutations, including T790M, V769L, and N771T.15, 67 The most important mutation in exon 20 is T790M, which is associated with a small fraction of adenocarcinomas with primary resistance to EGFR TKI and over one-half of the patients with acquired resistance to EGFR TKI (Figure 2).12, 67, 68, 69, 70, 71

Frequency of mutations in exons 18–21 of the EGFR gene and the association with responsiveness to EGFR targeted therapy. The EGFR located in chromosome 7p11.2 contains 28 exons. Exons 18–21 in the tyrosine kinase region of the EGFR gene are scaled up; a detailed list of EGFR mutations in these exons associated with sensitivity (green) or resistance (orange) to EGFR TKI.6, 12, 67, 68, 69, 70, 71, 80, 81, 82, 83, 84, 195 The frequency of the mutations is labeled to the side of the color-coded bars. The most prevalent EGFR mutations are in-frame deletions of exon 19 (45%), followed by L858R substitution in exon 21 (41%). Exon 18 mutations (G719A/C/S) account for ∼5% of the overall mutations. The exon 19 deletions, L858R in exon 21, G719A/C/S in exon 18, the L861Q and L861R in exon 21, are mutations that predict the probability of benefit from EGFR TKI therapy of adenocarcinomas. The insertion mutations in exon 20 (D770_N771 (insNPG), D770_N771 (insSVQ), D770_N771 (insG)) are the second most common and are associated with EGFR TKI therapy resistance. D761Y in exon 19 is also associated with resistance to EGFR TKI although it occurs in low frequency. *T790M mutation represents ∼1% of primary resistance but over 50% of acquired resistance in adenocarcinomas. **There are more than 20 exon 19 deletion forms in the lung adenocarcinomas, with the most common ones including delE746-A750, delL747-T751linsS, and delL747–P753insS.
A comprehensive literature review by Yamamoto et al33 indicated that 569 mutations were found in 2880 lung cancer patients (20%). The distribution of EGFR mutations was as follows: 48% in exon 19, 43% in exon 21, 4% in exon 20, and 3% in exon 18. EGFR mutations, except EGFRvIII, are rarely found in squamous cell and large cell carcinomas, thus EGFR TKI therapy may not be a relevant therapy for patients with those tumors. In a large series of lung carcinomas investigated for the presence of EGFR mutations in exons 18, 19, and 21, no EGFR mutations were found in the 454 squamous carcinomas and 31 large cell carcinomas investigated. In contrast, EGFR mutations were found in 10% of 375 adenocarcinomas and in 26% of the 86 cases designated as bronchioloalveolar carcinomas.27
The most commonly used method to detect EGFR mutations is direct sequencing.23, 25 It is noteworthy that tissue slides frequently contain heterogeneous components of cells, a fact that sometimes hampers optimal analysis. In addition, some patients present with multifocal lung tumors.72 Careful dissection of cells from a suitably representative area selected by a pathologist is essential to ensure a successful test result. Other methods include PCR–single-strand conformational polymorphism analysis73, 74 and high resolution-melting amplicon analysis.75, 76 Relative to the direct sequencing method, the other two techniques allow for the rapid detection of EGFR mutations with high sensitivity and specificity. However, confirmation of mutations via direct sequencing is still necessary.27, 76, 77 Though not of any current clinical use, an assay that provides a rapid assessment of EGFR mutation status in as little as 30 min using a ‘smart amplification process’ has been described. These may one day provide greatly improved turnaround times for this analysis.78 Formalin-fixed and paraffin-embedded tissue is perfectly suitable for fluorescence in situ hybridization (FISH) and DNA-based tests, but tissue preservation is critical for a successful test. Decalcified and ethanol-fixed tissue, as well as tissues containing abundant necrosis, should be avoided.
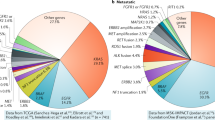
The ability to detect multiple driver mutations in lung adenocarcinoma has revolutionized the medical management of this disease and multiplexed testing for all common driver mutations will provide physicians with a more precise guide for therapy.9 Recently, Kris et al79 identified 10 driver mutations in tumor samples from 1000 lung adenocarcinoma patients enrolled in the National Cancer Institute Lung Cancer Mutation Consortium. The mutations, involving KRAS, EGFR, ERBB2 (HER2), BRAF, PIK3CA, AKT1, MAP2K1, and NRAS, were screened using standard multiplexed assays and FISH. Driver mutations were detected in 60% of tumors. The incidences of mutations were as follows: KRAS 25%, EGFR 23%, ALK rearrangements 6%, BRAF 3%, PIK3CA 3%, MET amplifications 2%, ERBB2 1%, MAP2K1 0.4%, NRAS 0.2%, and AKT1 0% (Figure 3).12, 67, 68, 69, 70, 71 It is noteworthy that 95% of molecular lesions were mutually exclusive.79

Frequency of major driver mutations in signaling molecules in lung adenocarcinomas. About 64% of all adenocarcinoma cases harbor somatic driver mutations. According to the National Cancer Institute Lung Cancer Mutation Consortium data,79 ∼23% of lung adenocarcinomas harbor EGFR mutations. The EGFR mutation status of the cancer is associated with its responsiveness or resistance to EGFR TKI therapy. KRAS mutations are more frequently found in adenocarcinomas (25%), which are mutually exclusive with EGFR mutations. Mutations in KRAS have been proposed as one of the mechanisms of primary resistance to gefitinib and erlotinib therapy. A subset of adenocarcinoma cases harbors a transforming fusion gene, EML4–ALK (6%), which mainly involves adenocarcinoma from non-smokers with wild-type EGFR and KRAS mutations. The mutation frequency of BRAF is 3%, PIK3CA 3%, MET amplifications 2%, ERBB2(Her2/neu) 1%, MAP2K1 0.4%, and NRAS 0.2%. Each of the molecular alterations has a role in the signal pathways, activating important cell functions, including cell proliferation and survival. Approximately 36.4% of lung adenocarcinomas do not harbor currently detectable mutations.
EGFR mutations are responsible for the constitutive activation of the tyrosine kinase receptor. These mutations are also most frequently associated with either sensitivity or resistance to EGFR TKIs (Figure 2).6, 80, 81, 82, 83, 84 The response-associated mutations are linked with response rates of >70% in patients treated with either erlotinib or gefitinib.85, 86 However, upto 25% of patients with TKI resistance-associated mutations will also respond to the therapy.67 Pao et al7 analyzed EGFR mutation of exons 18–24 in tumors from 10 gefitinib-responsive and from 7 erlotinib-responsive patients. The results demonstrated that EGFR mutations were present in 7 of 10 (70%) gefitinib-responsive and in 5 of 7 (71%) erlotinib-responsive tumors.
EGFR genotype was more useful than clinical characteristics for selection of appropriate patients for consideration of first-line therapy with an EGFR TKI.85 EGFR mutations are generally associated with sensitivity to TKI therapy.71, 87 Both retrospective and prospective studies have demonstrated that lung adenocarcinoma patients carrying such an EGFR mutation and who were treated with TKIs had significantly higher response rates and longer progression-free survival than patients without an EGFR mutation,5, 6, 7, 25, 29, 71, 83, 85, 87, 88 with some patients experiencing rapid, complete, or partial responses that were persistant.55 Jackman et al85 studied 223 chemotherapy-naïve patients with advanced lung cancer of non-small cell type, among which 86% were adenocarcinomas. Sensitizing EGFR mutations were found in 84 carcinomas, 89% of which were adenocarcinomas. The mutations were associated with a 67% response rate, with a time to progression of 11.8 months, and overall survival of 23.9 months.85 Exon 19 deletions were associated with a relatively longer median time to progression and overall survival compared with L858R (exon 21) mutations. Wild-type EGFR was found in 139 patients (62%), and this finding was associated with poor outcomes (response rate, 3%; time to progression, 3.2 months), irrespective of KRAS status.
EGFRvIII Mutation
EGFR variant III (EGFRvIII), a mutation resulting from an in-frame deletion of exons 2–7 of the coding sequence (amino acids 6–273), has been associated with a subset of squamous cell lung cancers.89, 90, 91 A number of functional differences between EGFRvIII and EGFR have been characterized.90, 91 EGFRvIII has been identified in an array of human solid tumors, including glioblastoma, breast cancer, ovarian cancer, prostate cancer, and lung caner. Although EGFRvIII fails to bind EGF, its intracellular tyrosine kinase is constitutively activated, allowing the receptor to undergo tyrosine autophosphorylation.92, 93, 94 EGFRvIII activates the phosphatidylinositol 3′ kinase (PIK3CA) signaling pathway, which provides critical information for cell survival, proliferation, and motility.95, 96 The true incidence and clinical significance of EGFRvIII mutations are not yet clearly defined. There have also been reports that EGFRvIII mutation in lung cancer correlates with increased EGFR copy number.97
Ji et al98 determined that EGFRvIII mutations were present in 5% (3/56) of human lung squamous cell carcinomas, but not in adenocarcinomas (0/123). The information concerning whether EGFRvIII mutation is associated with specific histological types of lung cancer is conflicting, although most studies have indicated that EGFRvIII is associated with squamous cell carcinoma.98, 99 In the study by Sasaki et al,97 EGFRvIII mutation was detected in 3% (8/252) of non-selected lung cancer patients. All patients bearing a EGFRvIII mutation were male, smokers, and seven had squamous cell carcinoma, whereas one had poorly differentiated adenocarcinoma. However, in the investigation of Ohtsuka et al,99 EGFRvIII mutation was detected in one of seven squamous cell carcinomas with an adenocarcinoma component, in two of four adenosquamous carcinomas and in one of seven large cell carcinomas.
EGFRvIII-bearing squamous cell carcinomas were reportedly insensitive to gefitinib and erlotinib, but showed sensitivity to neratinib (HKI-272).98, 100
EGFR Copy Number Alteration
Some, but not all, studies have shown that EGFR gene amplification is associated with significantly better survival after treatment with TKI.10, 30, 101 Despite the fact that the majority of studies demonstrate that a high EGFR gene copy number correlates with better response and increased survival in adenocarcinoma patients treated with EGFR TKI, debate remains about its true prognostic value. Dahabreh et al31 reviewed 59 publications concerning 1020 mutations in 3101 patients. EGFR mutations were detectable in 70% (15/21) of patients who had a gain of EGFR copy number. There are several methods for detecting and determining EGFR gene copy number, or dosage, including FISH,25, 30, 102 chromogenic in situ hybridization,32, 103 and real-time quantitative PCR.10, 104, 105 When compared with EGFR mutations, EGFR gene copy number gain was a less sensitive and less specific marker, and may therefore not be considered clinically suitable for patient selection.31
EGFR Protein Overexpression
There are three main types of immunohistochemical tests for EGFR protein: total EGFR, phosphorylated EGFR, and mutant-specific EGFR.
Total EGFR
Overexpression of total EGFR has been demonstrated in 40–80% of tumors representing various subtypes of lung tumors; however, the use of EGFR overexpression as a prognostic marker has been largely unsuccessful.106, 107, 108, 109 Many studies suggest that immunohistochemistry-based assays measuring EGFR expression do not serve as a robust predictors of response to TKI therapy.110 The study from Li et al111 further emphasized that EGFR overexpression appears to be independent of EGFR mutation. As total EGFR did not correlate well with EGFR mutations, it is not accepted as a marker for EGFR TKI treatment selection.
Phosphorylated Form of EGFR
Phosphorylations in the carboxyl-terminus of EGFR have a key role in the recruitment of signaling molecules and activation of downstream signaling pathways.51, 112, 113 The utility of detecting phosphorylated EGFR remains questionable owing to concerns about its stability and its compatibility with routine pathology practice. Further studies are warranted to evaluate the potential clinical utility of antibodies that recognize phosphorylated EGFR.
EGFR Mutation-Specific Antibodies
The current commercially available antibodies recognize two of the most common EGFR mutations ((delE746_A750) in exon 19 and L858R in exon 21).114 The antibodies successfully detected EGFR alterations in 51 of 217 adenocarcinomas and in 1 of 217 squamous carcinomas. These findings were confirmed by DNA sequencing.114 Immunohistochemistry using mutation-specific antibodies could potentially be used to screen for patients who may be candidates for EGFR inhibitors.115 However, there are concerns about the limited mutation types the antibodies recognize and a practical cutoff point for deeming a test positive or negative has yet to be established. In light of currently available data, it has been proposed that commercially available antibodies may be most useful for initial screening.
Other Alterations that Affect the EGFR TKI Response
Other gene mutations downstream of the EGFR signaling pathway are also involved in tumorigenesis of lung adenocarcinomas. Studies have indicated that any activating mutation in the EGFR/RAS/RAF1 signaling pathway may be sufficient for the pathogenesis of certain lung cancers.116, 117 The findings of Yamamoto et al33 indicated that EGFR-mutant cancers tend to have fewer downstream molecular alterations. If EGFR-mutant cancers acquire other critical molecular alterations, genetically or epigenetically, they may survive by using alternative signaling pathways, even if the EGFR signaling is effectively inhibited. It is expected that in the presence of additional molecular alterations, the effectiveness of TKI therapy for EGFR-mutant tumors will be reduced. The most frequently encountered alterations include KRAS mutations,118, 119, 120, 121 MET amplification,122, 123, 124, 125 ALK gene fusion,126, 127, 128, 129 PIK3CA mutations,130 BRAF mutations,118, 131, 132, 133 and IGF1R overexpression (Figure 3).134, 135
Is EGFR mutation specific for lung adenocarcinoma?
Current data indicate that EGFR mutations are adenocarcinoma dominant, rather than adenocarcinoma specific. Clinically, most EGFR mutations are detected in adenocarcinomas;27 with other types of lung carcinomas showing a much lower frequency of EGFR mutations: 5% in squamous cell carcinomas and virtually none in large cell carcinomas.27, 136 Lung adenocarcinomas frequently possess EGFR mutations and frequently exhibit increased EGFR copy number.111 A study including 334 cases of lung adenocarcinoma using PCR-based assays to detect deletions within exon 19 and the L858R mutation in exon 21 of the EGFR gene found that 23% of these tumors contained a mutation. Of those, 29% were exon 19 deletions and 29% were the L858R mutation in exon 21. In addition, EGFR amplification, defined as greater than five EGFR signals per nucleus, was detected in 52% of EGFR-mutated tumors, but in only 6% of those lacking the EGFR mutations.111 Among adenocarcinomas, EGFR mutations are more prevalent in cases formerly subtyped as bronchioloalveolar carcinomas.27, 41, 99 However, EGFR mutations in squamous cell carcinoma appear to occur much less frequently, with a reported incidence as high as 14% from a group of seven patients to as low as 0% in a group of 454 squamous cell lung cancers.27, 41, 99 The National Comprehensive Cancer Network recommends erlotinib as the first-line therapy for patients who have an EGFR mutation and who have advanced, recurrent, or metastatic adenocarcinoma.3
Adenosquamous carcinomas appear to have an EGFR mutation incidence that is similar to that of adenocarcinomas. Furthermore, adenosquamous carcinomas have been treated in a manner similar to adenocarcinomas as these tumors harbor comparable EGFR mutations. In a study that included 23 adenosquamous carcinomas with separately microdissected glandular and squamous components analyzied for EGFR and KRAS mutations, EGFR mutations were observed in 13% of cases (3/23), two of which had identical mutations in the glandular and squamous elements.137
Although EGFR mutations in lung cancers other than adenocarcinoma type are not common, occasionally reported cases bearing EGFR mutation have indicated that EGFR TKI therapy could still be a potentially effective approach for those patients (Figure 4). Large cell carcinoma, appears to harbor EGFR mutations very rarely.27, 138, 139 In one study, only one L858R mutation was found in 60 large cell carcinomas of lung.139 In the study of Marchetti et al,27 EGFR mutations were identified in 39 (10%) of 375 adenocarcinomas, but no EGFR mutations were found in 454 squamous carcinomas and 31 large cell carcinomas. De Pas et al140 reported a 66-year-old woman with metastatic large cell lung cancer harboring EGFR mutation. A positron emission tomography scan performed 2 months after the initiation of gefitinib therapy showed a dramatic response to treatment in both the patient's primary tumor and her metastatic deposits.

Current molecular tests and options for targeted therapies. Adenocarcinomas and adenosquamous carcinomas have a relatively high incidence of EGFR mutations or EML4–ALK rearrangement. Patients with such tumors could potentially benefit from targeted therapies using EGFR TKI and ALK TKI. EGFRvIII is associated with a small subset of squamous cell carcinomas, but the rationale for the therapy targeted to this mutation has not yet been established. Large cell and sarcomatoid carcinomas are not considered suitable tumors for the EGFR TKI therapy although a recent article reported 28% EGFR mutations from a group of 32 sarcomatoid lung cancers.
Sarcomatoid lung cancer is rare and highly malignant. Whether EGFR mutation is involved in its tumorigenesis is unclear. Jiang et al141 investigated a group of 33 patients with sarcomatoid lung cancer for EGFR mutations by direct sequencing. EGFR mutations were detected from 9 of 32 patients and only one patient had a KRAS mutation.
In contrast to the EGFR mutations that are often found in adenocarcinomas, pure squamous cell carcinomas exhibit mutations in the discoidin domain receptor tyrosine kinase 2 with a frequency of 3.8% (11/290).142 Discoidin domain receptor 2, a tyrosine kinase receptor that binds collagen as its endogenous ligand, has been previously shown to promote cell migration, proliferation, and survival when activated by ligand binding and phosphorylation. EGFRvIII mutations were found in 5% of human lung squamous cell carcinomas (also see prior discussion) (Figure 4).98, 99
The advent of targeted therapy based on driver mutations in lung adenocarcinoma has countered the notion that non-small cell lung cancer (NSCLC) is a distinct clinical entity. Current information indicates that distinguishing a tumor as NSCLC alone is no longer sufficient for patient management and the term ‘non-small cell lung cancer (NSCLC)’ should be abandoned. Recently, a panel of experts proposed a major revision of the lung cancer classification system.143 These changes primarily affect the classification of adenocarcinoma and its distinction from squamous cell carcinomas (Table 1).143 The new classification system from the International Association for the Study of Lung Cancer, the American Thoracic Society, and the European Respiratory Society further classifies lung carcinomas into more precise subtypes based on a multiparameter approach that incorporates and integrates clinical, molecular, and histological features (Table 1).143 The advent of mutation-specific therapies has dramatically changed the landscape of lung cancer treatments.3, 13 Patients without EGFR mutations seldom respond to EGFR-targeted therapy. Targeted therapy drugs are inherently costly as these carefully designed molecules are patented, yet aimed at a limited patient population. Inappropriate use of these and other targeted therapies run the risk not only of clinical failure, but also of unnecessary expense or even life-threatening toxicity.
EML4–ALK rearrangement: driver mutation of lung cancer
The ALK gene encodes a receptor tyrosine kinase found in a number of fusion proteins consisting of the intracellular kinase domain of ALK and the amino terminal portions of different genes.144, 145 EML4–ALK fusion is formed as the result of a small inversion within the short arm of chromosome 2 that joins intron 13 of echinoderm microtubule associated protein-like 4 (EML4) to intron 19 of ALK [inv(2)(p21;p23)], generating an oncogenic fusion encoding a constitutively activated protein tyrosine kinase.8, 146 A subset of lung adenocarcinoma cases harbor within the genome this transforming fusion gene, EML4–ALK (Figure 5).

Schematic of EML4–ALK rearrangement, its detection by FISH, and its downstream effects. Both EML4 and ALK genes are located on the short arm of chromosome 2. The EML4–ALK rearrangement results from a chromosomal inversion, t(2;5) (a). Green and orange bars represent DNA probes corresponding to the 5′ and 3′ fragments of the ALK gene. The EML4–ALK fusion gene is mainly found in adenocarcinomas that arise in non-smokers with wild-type EGFR and KRAS. The EML4–ALK fusion protein activates canonical signaling pathways, including STAT3, RAS/MAP2K1, and PIK3CA/AKT1 cascades, which further affect cell cycle regulation, cell proliferation, neovascularization, and cell survival. At least nine variants have been identified. FISH detection of ELM4–ALK uses break-apart technology, which detects the adjacently located EML4 and ALK genes in wild-type signals (overlapping green-red) (b), and break-apart signals (separated green-red in one set of green-red) caused by chromosomal inversion (c).
The EML4–ALK fusion is a rare abnormality detected in ∼3–13% of patients with adenocarcinomas.8, 146, 147, 148, 149, 150, 151, 152 The most common fusion results from the joining of exons 1–13 of EML4 to exons 20–29 of ALK. At least seven EML4–ALK variants (V1–V7) have been identified in lung adenocarcinomas.153 All seven variants are formed through the fusion of the intracellular tyrosine kinase domain of ALK with a variably truncated EML4 gene.8, 154, 155, 156 Activated ALK is involved in the inhibition of apoptosis and the promotion of cellular proliferation through activation of downstream PIK3CA/AKT1- and MAPK1-signaling pathways.157 Fusion of the EML4–ALK gene and its associated EML4–ALK product may further lead to constitutive activation of the RAS/RAF1/MAP2K1/MAPK1 pathway.54 Additionally, two other less frequent ALK fusions in lung cancer have been reported, but not yet studied for their downstream consequences.158 The key downstream effectors on the ALK pathway include the RAS-activated protein, extracellular signal regulated kinase (MAPK1), phosphoinositide 3-kinase (PIK3CA), and STAT3 signaling pathways.151 RAS/MAP2K1/MAPK1 pathways are critical for cell proliferation, whereas the PIK3CA/AKT1 and STAT3 pathways are important for cell survival. The histology of these tumors is typically characterized by mucin production and either a solid growth pattern containing signet ring cells in western patients or an acinar growth pattern in Asian patients.159, 160, 161, 162 Compared with patients with wild-type ALK and EGFR, patients with the EML4–ALK fusion gene tend to be younger (median, 52 vs 64 years), of Asian ethnicity, diagnosed at an advanced clinical stage at presentation, male dominant (58 vs 32%), and more likely to be never-smokers (74 vs 26%), but with a comparable response rate to chemotherapy and overall survival.8, 148, 159, 160 The EML4–ALK fusion gene was detected in 19 of 141 (13%) tumor samples by FISH.148 None of the 10 patients with an EML4–ALK rearrangement achieved an objective response to EGFR TKIs. In contrast, 24% of responding patients in the EML4–ALK-negative cohort showed an objective response with an EGFR TKI.148, 163 Tiseo et al126 reported a 48-year-old Caucasian never-smoker man with lung adenosquamous carcinoma harboring EML4–ALK fusion and exon 19 deletion in EGFR gene. The patient manifested resistance to the erlotinib therapy. The authors concluded that ALK status should be investigated in unexplained cases of EGFR TKI-resistance lung cancers. In the study of Shaw et al,148 none of the 10 patients with EML4–ALK fusion had a documented clinical response to erlotinib. As the presence of the EML4–ALK fusion gene is mutually exclusive with the EGFR mutation, it is unclear if EGFR TKI resistance is owing to EML4–ALK mutation itself or because of the EGFR wild-type.
It has been reported that although ALK-fusion-positive lung cancers are resistant to the EGFR TKIs, gefitinib, and erlotinib, they are sensitive to small molecule TKIs against ALK.152 ALK TKIs (ALK TKI), including crizotinib, are effective treatments in preclinical models for patients with ALK-fusion cancers.164 In a pivotal phase 1 clinical trial, the ALK tyrosine kinase inhibitor (TKI) crizotinib (PF-02341066) demonstrated impressive antitumor activity in the majority of patients with adenocarcinomas harboring EML4–ALK fusions. However, despite these remarkable initial clinical responses, these cancers eventually developed resistance to crizotinib, usually within 1 year, thereby limiting the potential clinical benefit of this drug. Katayama et al165 found that cells resistant to intermediate doses of crizotinib develop either amplification of the EML4–ALK gene or a gatekeeper mutation, L1196M, within the kinase domain. Sasaki et al164 proposed two mechanisms of ALK TKI resistance based on evidence from a crizotinib-treated ALK-positive lung cancer patient and in a cell line generated from the resistant tumor. The crizotinib-resistant DFCI076 cell line harbored a unique L1152R ALK secondary mutation and was also resistant to the structurally unrelated ALK TKI, TAE684. In contrast, the TAE684-resistant (TR3) H3122 cell line did not contain an ALK secondary mutation, but instead harbored coactivation of EGFR signaling. Therefore, the authors suggested that dual inhibition of both ALK and EGFR was the most effective therapeutic strategy for the DFCI076 and H3122 TR3 cell lines.164
In a recent analysis of 82 ALK-positive lung cancer patients, ALK tyrosine kinase inhibitor (crizotinib) therapy was associated with improved survival compared with that of crizotinib-naive controls.166 Survival among ALK-positive patients who were given crizotinib in the second-line or third-line setting was significantly better than those patients given any second-line therapy (2-year survival, 55 vs 12%). However, unlike EGFR mutation, ALK rearrangement was not a favorable prognostic factor.166
Dual-color–split-apart FISH is the recommended method for the EML4–ALK test. A positive test result in >15% of 50 analyzed tumor cells is the cutoff point recommended by the College of American Pathologist and the Association of Molecular Pathology. The results of ALK FISH interpretation should be verified by two independent personnel. RT-PCR is not recommended due to repeated failures of RNA-based testing using formalin-fixed paraffin-embedded tissue. Immunohistochemistry is also not currently recommended as an alternative to FISH testing, due to the low expression level and resulting suboptimal sensitivity and specificity.167, 168
EGFR targeted therapy approaches
There are two major approaches for inhibiting EGFR signaling: (1) prevent ligand binding to the extracellular domain with a monoclonal antibody and (2) inhibit the intracellular tyrosine kinase activity with a small molecule TKI. Ligand binding to the extracellular domain of EGFR promotes receptor dimerization, which in turn leads to activation of the cytoplasmic tyrosine kinase. The activated EGFR kinase phosphorylates tyrosines in the EGFR C-terminal, initiates signaling cascades, and stimulates cell growth and differentiation (Figure 6).13, 169, 170

Mechanism of constitutive activation of EGFR results from EGFR mutation and strategies of anti-EGFR therapy. (a) EGFR mutations provoke autophosphorylation of key tyrosine residues (P) in the tyrosine kinase domain, thus activating tyrosine kinase activity constitutively and initiating downstream effectors. (b) Two strategies are used for inhibiting EGFR signaling: humanized antibodies and small molecule TKIs. The antibodies inhibit the ligand-dependent activation of EGFR by blocking the ligand-binding site and preventing EGFR from activation. In contrast, TKIs block the magnesium-adenosine triphosphate-binding pocket of the intracellular tyrosine kinase domain, further inhibiting autophosphorylation. This inhibition disrupts tyrosine kinase activity and abrogates intracellular downstream signaling.
Monoclonal Antibodies
Monoclonal antibodies, such as cetuximab and panitumumab, are either chimeric mouse-human or fully humanized antibodies targeting the EGFR extracellular domain, leading to blockade of ligand-activated signal transduction and receptor dimerization. Fully humanized antibodies, such as panitumumab, have a high affinity for EGFR and a longer half life.171
The binding of the antibody initiates EGFR internalization and degradation, which leads to signal termination.110, 172, 173, 174 The treatment has shown consistent benefit to clinical outcome when added to chemotherapy.173 However, this class of treatment only inhibits ligand-dependent activation of EGFR and not autophosphorylation of the tyrosine kinase domain via constitutive activation. These mutations may still activate the downstream pathways, and upregulate cell cycle progression, cell growth, and angiogenesis.
Tyrosine Kinase Inhibitors
TKIs are synthetic small molecules that block the magnesium-ATP-binding pocket of the intracellular tyrosine kinase domain.110 TKIs prevent the intracellular tyrosine kinase domain of the EGFR from autophosphorylation through binding to its ATP-binding site. Several TKIs, such as gefitinib and erlotinib, are specific for EGFR, whereas others inhibit other receptors in addition to EGFR, such as ERBB2 and VEGFR2. TKIs block ligand-induced receptor autophosphorylation by binding to the tyrosine kinase domain and disrupting tyrosine kinase activity, thereby abrogating intracellular downstream signaling. Somatic activating mutations of the EGFR gene in exons 19 and 21 increase gene copy number. Certain clinical and pathological features have been associated with dramatic tumor responses and favorable clinical outcomes with these agents in patients with lung cancer.30, 55, 175
Mechanisms of Resistance to EGFR Targeted Therapy
In a population of unselected patients, response to EGFR TKIs has been reported to be only 10%.176 These disappointing outcomes may reflect the fact that the majority of lung cancers are EGFR-mutation negative. Moreover, the outcomes may also be related to evidence that some EGFR mutations not only activate EGFR tyrosine kinase and drive cancer cells to grow, but are also associated with resistance to current EGFR TKI therapy.82, 84, 177, 178 Current strategies have been focused on detection of EGFR mutation and EML4–ALK rearrangement in lung adenocarcinomas (Figure 7).

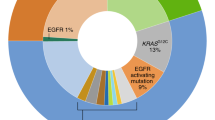
Suggested algorithm for molecular testing for patients with lung adenocarcinoma. The algorithm defines the rationale in selecting patients who could benefit from EGFR and EML4–ALKA targeted therapy. Adenocarcinoma cases are subjected to testing for EGFR mutations. The EGFR mutation-positive cases (25%) are further divided into responsive and resistant groups according to their mutation profiles. A responsive mutation predicts a response rate of 91% and a resistant mutation predicts a response rate of 9%. The presence of wild-type EGFR characterizes about 75% of the adenocarcinomas, and predicts the likelihood of non-responsive to EGFR TKI. Tumors with wild-type EGFR are further tested for EML4–ALK rearrangement. Although EML4–ALK rearrangement is found in only 3% of patients with lung adenocarcinoma, its presence predicts a 53% probability of response to targeted therapy.
The most important mutation associated with acquired EGFR TKI resistance is T790M, a point mutation located at exon 20, resulting in the substitution of methionine for threonine.70, 179, 180, 181 Clinically, patients with EGFR exon 20 mutations do not respond to gefitinib.67 Moreover, the appearance of a secondary mutation in exon 20 (T790M) accounts for ∼50% of acquired drug resistance.71, 182 Screening for the emergence of such mutations on circulating tumor cells from the blood of patients during the course of treatment may allow earlier identification of acquired resistance.178, 180 Other TKI-resistant mutations include insertions in D770 at exon 20 and D761Y at exon 19 (Figure 2).
Results of some preclinical studies suggest that the clinical benefit observed with EGFR TKIs is not restricted to those patients harboring EGFR gene mutations. This may be due to molecular factors other than EGFR mutation. EGFR gene amplification and receptor/ligand overexpression, both allowing for a ‘gain of function’ to occur, may account for some cases of tumor sensitivity to single-agent EGFR inhibitors.30, 36 Recent evidence suggests that the approach to accurately predicting response to TKI therapy in adenocarcinomas should combine the status of EGFR mutation and its copy number. Although multiple parameters have been used to predict tumor responsiveness to TKI, EGFR mutation detection remains one of the most important determinants for the prediction of clinical responsiveness and survival benefit.183
The EGFR pathway has a central role in a subset of adenocarcinomas through converging signals for cell proliferation, motility, and other cancer cell behaviors.5, 184 However, the mechanisms underlying tumor resistance to EGFR-targeted therapy are still not completely known. Genetic alterations frequently occur during lung cancer progression owing to genomic instability. Biological stress may also modulate multiple signaling pathways and trigger epigenetic alterations. Despite impressive initial clinical responses, patients with EGFR-mutated adenocarcinomas almost inevitably develop drug resistance after ∼1 year of TKI treatment.185, 186 Studies have revealed several molecular mechanisms that may contribute to the development of tumor resistance to TKI therapy,71, 84, 186, 187 including acquired secondary EGFR mutation, activation of alternative signaling pathways that bypass the EGFR pathway, overexpression of HGF,122 tyrosine protein kinase MET amplification,53, 124 epigenetic factors,188 constitutive activation of signaling pathways downstream of EGFR,13, 189 tumor stromal and extracellular matrix alterations,190, 191 or host-related mechanisms such as rapid drug inactivation and ATP-binding cassette transporters efflux.192
Primary resistance to EGFR targeted therapy
Recent clinical trials of gefitinib or erlotinib therapy report response rates in EGFR-mutated lung cancer cases ranging from 75 to 90%.13, 15, 22, 38, 193 A subset of lung adenocarcinomas show primary resistance to EGFR TKI therapy, even in the presence of an activating mutation in EGFR.194 The most commonly found mutations associated with TKI drug sensitivity include exon 19 deletions downstream of the lysine residue at position 745 (ΔE746-A750), point mutations in exon 21 (L858R and L861Q and L861R), in exon 18 (G719A/C/S), and in exon 20 (V765A, T783A, and S768I).67, 68, 69, 70, 71 However, insertion mutations of exon 20, D770_N771 (insNPG), D770_N771 (insSVQ), D770_N771 (insG), and point mutations in exon 20 (V769L, N771T) were associated with EGFR TKI resistance.12, 195 This observation has been confirmed in an in vitro model in which insertion mutations in exon 20 rendered transformed cells less responsive to EGFR TKIs compared with the sensitizing mutations of exons 19 and 21.195 Insertion mutation in exon 20 at 770 renders the EGFR 100-fold less sensitive to TKIs when compared with the sensitizing mutations.195 The mutation T790M, which is associated with ∼50% of acquired resistance, has also been linked to primary resistance, although infrequently (<5% of such cases) (Figure 2).179
Approximately 10–25% of EGFR-mutant lung adenocarcinomas do not respond to an EGFR TKI.15 In addition to the previously mentioned mutations, even rarer primary mutations, such as D761Y, G719C, and E709A mutations, have also been shown to be insensitive to EGFR TKIs, and even more so when co-occurring with other genetic alterations.196
Primary TKI resistance may also be mediated by the presence of other genetic alterations that affect signaling downstream from EGFR, such as mutation of KRAS, PIK3CA, and loss of PTEN expression.197, 198, 199 Mutations in KRAS, which are frequently found in adenocarcinomas with wild-type EGFR, are a mechanism of primary resistance to gefitinib and erlotinib.63 PTEN is one of the key downstream components of the EGFR pathway and has a significant role in cell survival, proliferation, and growth. Knockdown of PTEN expression in cells results in drug resistance to gefitinib and erlotinib.200 Loss of PTEN expression results in overactivation of the Akt pathway and confers resistance to EGFR TKI.198 Study results suggest that the loss of expression of PTEN may be mediated by an epigenetic mechanism, as genetic alterations on the PTEN gene are found in fewer than 10% of cases.201, 202 BRAF mutations may also be associated with primary resistance to EGFR TKIs.203
Wild-Type EGFR
The wild-type EGFR appears to be a significant marker for the primary EGFR TKI resistance. The Iressa Pan-Asia Study clinical trial demonstrated that most tumors without detectable EGFR tyrosine kinase domain mutations were insensitive to gefitinib.41 Tumors with wild-type EGFR often harbor somatic mutations in other genes that affect the key pathways in lung adenocarcinoma. Thus, primary drug insensitivity is likely linked to the absence of drug-sensitizing mutations in EGFR and is more likely to be a result of mutations in other genes.15, 85 Further complicating matters are the findings that even in tumors with EGFR mutations, certain mutations appear to confer greater sensitivity to treatment than others.33 This emphasizes that predicting response to treatment is a complex process that is not completely understood at this time.
KRAS Mutations
One of the most important discoveries for the clinical management of colorectal carcinoma has been the association of mutations in KRAS and the usual failure of monoclonal antibodies targeting EGFR, such as panitumumab and cetuximab. The KRAS protooncogene encodes KRAS G-protein, which has a critical role in the RAS/MAPK1 signaling pathway downstream of many growth factor receptors, including EGFR. Some tumors harbor somatic mutations in exon 2 of KRAS that compromise the hydrolysis of RAS-bound GTP to GDP, resulting in constitutive activation of the RAS pathway.204 In the presence of a KRAS mutation, the EGFR pathway activation is not significantly inhibited by cetuximab or panitumumab, which acts upstream of the KRAS protein, diminishing the efficacy of the agents. This pathway is identical to the EGFR pathway targeted by TKIs in adenocarcinomas.
KRAS has a key role in the EGFR signaling network. An activating mutation of KRAS is present in ∼25–35% of TKI non-responsive cases.63 EGFR and KRAS mutations are rarely detected in the same tumor, suggesting that they may perform functionally equivalent roles in lung tumorigenesis.205, 206 KRAS mutation is a negative predictor of response to anti-EGFR monoclonal antibodies and is also an important mechanism of resistance to TKIs in lung adenocarcinomas.55 A meta-analysis by Linardou et al207 provided empirical evidence that somatic mutations of the KRAS oncogene are highly specific negative predictors of response to single-agent EGFR TKIs in advanced lung cancers, mostly adenocarcinomas. Mao et al121 reviewed 1470 lung cancers from 22 publications, of which 231 had KRAS mutations (16%). The mutations were more common in adenocarcinoma than in other histological types of lung cancer (26 vs 16%). The objective response rate of patients with mutant KRAS was 3% (6/210), whereas the objective response rate of patients with wild-type KRAS was 26%. As most KRAS mutations are detected in codons 12 and 13 of exon 2, an alternative-screening algorithm is to perform KRAS mutation analysis first, followed by EGFR mutation test if KRAS mutations are not found (Figure 8).

Alternative algorithm for molecular testing for patients with lung adenocarcinomas. Approximately 25% of lung adenocarcinomas harbor KRAS mutations, which predict non-response to EGFR TKI therapy. Of the remaining KRAS-negative lung adenocarcinomas, ∼20% harbor EGFR mutations, which are associated with responsiveness to EGFR TKI therapy. EGFR mutation-negative cases may benefit from additional testing for the EML4–ALK rearrangement, which will be helpful in selecting patients potentially eligible for ALK targeted therapy.
Mutations in KRAS are one mechanism of primary resistance to gefitinib and erlotinib.208 KRAS mutations are almost exclusively detected in codons 12 and 13 of exon 2, resulting in EGFR independent intracellular signal transduction activation. In the study of Eberhard et al,11 EGFR exons 18 through 21 and KRAS exon 2 mutations were investigated via sequencing in tumors of 274 patients. KRAS mutations were present in 21% of tumors, which were associated with significantly decreased time to progression and survival in patients treated with erlotinib plus chemotherapy.
KRAS mutations were detected in 21/215 (10%) of adenocarcinomas but were not found in 15 squamous cell carcinomas or 11 lymphoepithelioma-like carcinomas.196 Santis et al209 observed a similar pattern. In a study including 121 adenocarcinomas from African–American patients, KRAS mutations were compared with data from Caucasian patients (n=476).210 The KRAS mutations were found in 17% (21/121) of African–American patients compared with 26% (125/476) of Caucasian patients.210 KRAS mutations in adenocarcinoma are usually associated with wild-type EGFR and non-responsiveness to EGFR TKI therapy. Therefore, it is difficult to determine whether the resistance is due to the presence of mutated KRAS or to the absence of mutated EGFR. Mao et al121 reviewed 22 studies, finding that KRAS mutations were detected in 150 of 718 (21%) patients with lung cancer. Mutations were more common among adenocarcinomas than in other types of cancer (26 vs 16%). The objective response rate of patients to EGFR TKI with mutant KRAS was 3% (6/210), compared with 26% (287/1125) of patients without KRAS mutations. Having a wild-type KRAS is very important if benefit is to be derived from EGFR inhibition, but is not the sole determinant of this outcome as other mechanisms of resistance to EGFR inhibitors exist.
BRAF Mutation
The BRAF gene encodes a protein that has a key role downstream of KRAS in the cell signaling pathway activating important cell functions, including cell proliferation and survival.132, 133 Both KRAS and BRAF genes are part of the signaling cascade for the EGFR family proteins.211 The BRAF protein is a serine/threonine protein kinase that is activated by KRAS in a GTP-dependent manner.118 Mutant BRAF proteins have elevated kinase activity and can transform NIH3T3 cells.212 KRAS function is not required for the growth of cancer cell lines with BRAF mutations.212 Among 697 patients with lung adenocarcinoma, BRAF mutations were present in 18 (3%) of the patients. The BRAF mutation frequencies were V600E (50%), G469A (39%), and D594G (11%), in exons 15, 11, and 15, respectively.213 All patients with BRAF mutations were current or former smokers. The major BRAF functions are believed to be mediated by phosphorylation and thus activate the MAPK1, MAP2K1, and MAP2K2 pathways.118, 214 Mutations in BRAF have been shown to impair responsiveness to panitumumab or cetuximab in patients with colorectal carcinomas.215 BRAF mutations are found in 1–3% of lung cancers, most of which are adenocarcinomas.206, 212 BRAF mutations are found in a mutually exclusive pattern with KRAS mutations, suggesting that these genetic events activate a set of common downstream effectors of transformation. However, BRAF exon 15 mutations were tested on 96 paired samples of primary lung adenocarcinomas and corresponding locoregional lymph node metastases.133 BRAF mutations were observed in two patients with KRAS mutations demonstrating the possibility of both mutations in BRAF and KRAS occurring in the same tumor.
Mutations in the KRAS/BRAF pathway were recently shown to predict clinical response to MEK inhibition in lung adenocarcinoma.56, 216 The histological phenotype of BRAF mutant adenocarcinomas has not been well described, but was reported to be a mixed type adenocarcinoma with a high incidence of papillary (80%) and lepidic growth (50%) patterns.217
De Oliveira Duarte Achcar et al218 investigated 15 primary micropapillary lung adenocarcinomas for KRAS, EGFR, and BRAF mutations. BRAF was found in three (20%) of these cases. The tumors had diverse histological characteristics including mucinous, lepidic, acinar, and solid growth pattern.
The initial retrospective work on mutant BRAF's effect on EGFR-targeted therapy was performed on a cohort of 132 metastatic colorectal cancer patients.215 The results showed that none of the patients who experienced a response displayed BRAF mutations, whereas 11 of 79 (14%) non-responders carried a BRAF V600E allele.215 As BRAF mutations are mutually exclusive to EGFR and KRAS mutations, it is likely to be associated with lack of response to EGFR TKIs.138
Other Genomic Alterations
Genetic alterations other than EGFR, KRAS, and EML4–ALK alterations are relatively rare in lung adenocarcinomas and available data concerning such alterations are limited. However, these less common alterations may have significant clinical importance. Current literature indicates that 30–40% of EGFR TKI-resistant EGFR-mutated tumors do not carry secondary resistance mutations.123, 134, 200, 219 The role of oncogenic activation of EGFR downstream effectors such as KRAS, BRAF, PIK3CA, and PTEN in response to therapy has been discussed extensively in a series of studies.54, 62 The RAS/MAPK1 and PIK3CA/AKT1 pathways are the major signaling networks linking EGFR activation to cell proliferation and survival.220 Activating mutations in these downstream effectors of EGFR signaling could lead to resistance to EGFR inhibitors.221, 222, 223 The discovery of other molecular resistance aberrations, such as MET kinase amplification or mutations of EML4–ALK fusion, which cause constitutive activation of RAS/RAF1/MAP2K1, has provided further insight and validation into factors limiting the therapeutic efficacy of EGFR inhibitors.53, 148, 224 The mystery as to why all tumors harboring drug-sensitive EGFR mutations do not respond to treatment with EGFR TKI inhibitors is yet to be resolved. Clinical investigations have shown that the presence of other genetic alterations affecting signaling pathways downstream of EGFR may have a crucial role. Mutation of PIK3CA confers gefitinib resistance and loss of PTEN expression also contributes to erlotinib resistance.198, 225
Heterogeneity within cancer cell populations in the response to anticancer therapy is another factor to be considered.72 However, some studies demonstrate that heterogeneity is not likely a significant factor that affects the EGFR targeted therapy. Yatabe et al226 compared EGFR mutation patterns between primary and metastatic sites and between primary and recurrent tumors. There were no discordant mutation patterns among 77 paired primary and metastatic site samples or among 54 primary and recurrent tumor pairs.
A recent study evaluated whether abundance of EGFR mutations in tumors predicts treatment outcome in 100 cases of advanced lung cancer, among which 93 were adenocarcinomas.205 Of the 100 samples studied, 51 and 18 samples harbored high and low abundances of EGFR mutations, respectively, and 31 carried wild-type EGFR. Differences in overall survival and objective response rate in patients with high and low abundances of EGFR mutations were not significant.227 This study also pointed out that heterogeneity caused by factors other than EGFR mutation could also affect EGFR TKI response. Sharma et al228 reported detecting a small subpopulation of reversibly ‘drug-tolerant’ cells showing more than 100-fold reduced drug sensitivity. The drug tolerant phenotype is maintained via engagement of IGF1 receptor signaling. Treatment against IGF1 could selectively ablate the drug-tolerant subpopulation, thereby potentially improving the therapeutic effectiveness of EGFR TKI.228
MET Amplification
MET also contributes to primary and acquired resistance to EGFR TKIs.194, 229 MET is located on chromosome 7q21, which encodes the tyrosine kinase, hepatocyte growth factor receptor.230 Amplification of MET is associated with acquired resistance to EGFR TKIs through a mechanism termed kinase switch.134 Overall, MET amplification has been reported in about 20% of tumors from patients with acquired resistance.9, 231, 232 MET amplification occurs in both squamous cell carcinoma and adenocarcinoma.232 In vitro studies have shown that MET amplification is associated with increased concentrations of phosphorylated hepatocyte growth factor receptor.53 Amplification of MET correlated with a poor prognosis in a study of surgically resected lung cancers, including 241 adenocarcinomas, 139 squamous cell carcinomas, and 67 other types of tumors.231
Aberrant MET signaling may have a key role in the development of acquired resistance to therapy with an EGFR TKI.53 The clinical relevance of MET amplification has been investigated by examining tumor biopsies from patients who developed acquired resistance to gefitinib or erlotinib. The MET copy status was assessed in rebiopsy samples from 18 lung cancer patients at the time of secondary resistance development following an initial partial response.53 MET amplification was detected in four patients (22%). In another study, MET amplification was identified in 9 of 43 (21%) patients who had developed secondary resistance to an EGFR TKI in contrast to 2 of 62 (3%) patients with known sensitizing EGFR mutations who also had amplification of MET.233 The identification of MET amplification has led to the development of hepatocyte growth factor receptor-targeted TKIs.234 Clinical trials are underway with hopes that this may provide another form of targeted in therapy in EGFR TKI nonresponders.
ERCC1 Expression
ERCC1 (excision repair cross-complementing 1) is a DNA repair gene located at 19q13.32, which encodes a protein consisting of 297 amino-acid residues. Defects in ERCC1 are the cause of cerebro–oculo–facio–skeletal syndrome type 4. The ERCC1 enzyme has a key role in the nucleotide excision repair pathway, and also removes cisplatin-induced DNA adducts.235 The prognostic significance of ERCC1 was accessed by Simon et al236 with real time quantitative-PCR in surgical specimens from chemotherapy-naïve patients.
Investigational results of 1207 lung cancer patients by Gandara et al237, 238 on the relationship between EGFR mutation status and ERCC1 gene expression indicated that EGFR mutant cancers are more likely to be categorized as ERCC1 low and, therefore, platinum sensitive. Immunohistochemistry evaluation of ERCC1 expression in tumors from 130 patients revealed that ERCC1 was expressed in 10% of EGFR-mutated tumors and in 70% of EGFR wild-type tumors.238 Patients with low ERCC1 expression had a longer overall survival compared with the patients with high ERCC1 expression.239 Although most studies indicate a consistent association between ERCC1 expression level and responsiveness to cisplatin-based therapy, another study reported discordance of ERCC1 expression between primary and metastatic tumors. This discordance was found in about 40% (n=49) of cases, potentially indicating challenges in the clinical application of ERCC1.240
Acquired resistance to EGFR TKI
Published data indicate that 70–80% of mutation-positive adenocarcinomas are EGFR TKI sensitive, whereas response rates of tumors with wild-type EGFR are only 10–20%.28, 39, 41, 241, 242 There are two well-established mechanisms of acquired resistance: additional mutations in the EGFR gene, acquired during the course of treatment, which change the protein coding sequence; and amplification of other oncogene signaling pathways, such as those involving the RAS and MET oncogenes.84, 221, 222, 223
Kobayashi et al182 reported a gefitinib-resistant advanced adenocarcinoma patient who had a relapse after 2 years of complete remission with gefitinib. The DNA sequence of the EGFR at relapse revealed the presence of a second point mutation, resulting in threonine–methionine amino-acid change at position 790 of EGFR (T790M). Structural modeling and biochemical studies showed that this second mutation led to gefitinib resistance.182 The same mutation was confirmed by Pao et al208 through molecular analysis of EGFR in patients with acquired resistance to gefitinib or erlotinib. These gefitinib-resistant cases contain the same secondary mutation (T790M) in the kinase domain as those reported by Kobayashi et al.182 Codon 790 of EGFR is considered to be the ‘gatekeeper’ residue, which is an important determinant of inhibitor affinity in the ATP-binding pocket of EGFR.110 Substitution of this residue in EGFR with a bulky methionine may cause resistance by steric interference with binding of TKIs, including gefitinib and erlotinib.182, 208, 243 This mutation confers a survival advantage to the tumor and is selected while the patient is receiving anti-EGFR TKI treatment.82, 179 This secondary mutation is quite prevalent, being found in up to 50% of EGFR-mutant tumors treated with first-generation EGFR TKIs.182, 208
Arcila et al244 recently analyzed sensitizing EGFR mutations in 121 lung cancer patients, but only 104 (86%) cases were tested successfully. Most test failures were related to low-tumor content in the tested samples. All tested cases (61/61) with matched pretreatment and resistance specimens showed concordant sensitizing EGFR mutations. T790 M mutation was identified in 51 of 99 patients (52%) of cases analyzed. Retesting of 30 T790M-negative patients by the highly sensitive locked nucleic acid-based method detected 11 additional mutants for an overall prevalence of 68%.244
To address this particular cause of resistance, current third-generation EGFR TKIs are in development that irreversibly bind the ATP-binding socket in the presence of T790M.15 This compound, WZ4002, has a selective affinity for doubly mutated cells and, therefore, may represent a second-line treatment in cases of acquired resistance due to T790M. Further trials are needed to assess the potential efficacy of this approach before its clinical application. Because of the rarity of secondary mutations other than T790M in the EGFR tyrosine kinase domain, routine analysis of cases with acquired resistance is not recommended.
Conclusions
In summary, the advent of EGFR and ALK TKI therapy has provided a powerful new treatment modality for patients diagnosed with lung adenocarcinoma. Yet, primary and acquired resistance to targeted therapy continues to be a major obstacle to satisfactory clinical outcomes. Identification of the specific molecular alterations that contribute to response to EGFR targeted therapy will become a critical part of the process of selecting patients for appropriate treatments. Along with a growing understanding of the mechanisms of pharmacotherapy and the evolution of molecular resistance, it is anticipated that in order to maximize therapeutic effect and improve overall survival lung cancer treatments will be specifically tailored for the individual patient based on the presence or absence of critical molecular alterations.
References
Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
Siegel R, Ward E, Brawley O, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011;61:212–236.
Ettinger DS, Akerley W, Bepler G, et al. Non-small cell lung cancer: clinical practice guidelines in oncology. J Natl Compr Canc Netw 2011;8:740–801.
Travis WD, Brambilla E, Noguchi M, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 2011;6:244–285.
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497–1500.
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139.
Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004;101:13306–13311.
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–566.
Pao W, Girard N . New driver mutations in non-small-cell lung cancer. Lancet Oncol 2011;12:175–180.
Bell DW, Lynch TJ, Haserlat SM, et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol 2005;23:8081–8092.
Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005;23:5900–5909.
Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169–181.
Cheng L, Zhang S, Alexander R, et al. The landscape of EGFR pathways and personalized management of non-small-cell lung cancer. Future Oncol 2011;7:519–541.
Ciardiello F, Tortora G . EGFR antagonists in cancer treatment. N Engl J Med 2008;358:1160–1174.
Pao W, Chmielecki J . Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010;10:760–774.
Cheng L, Zhang D . Molecular Genetic Pathology. Humana Press/Springer: New York, New York, 2008.
Wang X, Zhang S, MacLennan GT, et al. Epidermal growth factor receptor protein expression and gene amplification in small cell carcinoma of the urinary bladder. Clin Cancer Res 2007;13:953–957.
Marks RA, Zhang S, Montironi R, et al. Epidermal growth factor receptor (EGFR) expression in prostatic adenocarcinoma after hormonal therapy: a fluorescence in situ hybridization and immunohistochemical analysis. Prostate 2008;68:919–923.
Wang X, Zhang S, Maclennan GT, et al. Epidermal growth factor receptor protein expression and gene amplification in the chemorefractory metastatic embryonal carcinoma. Mod Pathol 2009;22:7–12.
Cohen EE, Davis DW, Karrison TG, et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: a phase I/II study. Lancet Oncol 2009;10:247–257.
Wang X, Jones TD, Zhang S, et al. Amplifications of EGFR gene and protein expression of EGFR, Her-2/neu, c-kit, and androgen receptor in phyllodes tumor of the prostate. Mod Pathol 2007;20:175–182.
Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology Provisional Clinical Opinion: Epidermal Growth Factor Receptor (EGFR) Mutation Testing for Patients With Advanced Non-Small-Cell Lung Cancer Considering First-Line EGFR Tyrosine Kinase Inhibitor Therapy. J Clin Oncol 2011;29:2121–2127.
Sholl LM, Xiao Y, Joshi V, et al. EGFR mutation is a better predictor of response to tyrosine kinase inhibitors in non-small cell lung carcinoma than FISH, CISH, and immunohistochemistry. Am J Clin Pathol 2010;133:922–934.
Pao W, Miller VA . Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol 2005;23:2556–2568.
Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 2005;97:643–655.
Kosaka T, Yatabe Y, Endoh H, et al. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004;64:8919–8923.
Marchetti A, Martella C, Felicioni L, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol 2005;23:857–865.
Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol 2005;23:2513–2520.
Takano T, Ohe Y, Sakamoto H, et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol 2005;23:6829–6837.
Hirsch FR, Herbst RS, Olsen C, et al. Increased EGFR gene copy number detected by fluorescent in situ hybridization predicts outcome in non-small-cell lung cancer patients treated with cetuximab and chemotherapy. J Clin Oncol 2008;26:3351–3357.
Dahabreh IJ, Linardou H, Siannis F, et al. Somatic EGFR mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2010;16:291–303.
Sholl LM, John Iafrate A, Chou YP, et al. Validation of chromogenic in situ hybridization for detection of EGFR copy number amplification in nonsmall cell lung carcinoma. Mod Pathol 2007;20:1028–1035.
Yamamoto H, Toyooka S, Mitsudomi T . Impact of EGFR mutation analysis in non-small cell lung cancer. Lung Cancer 2009;63:315–321.
Tamura K, Okamoto I, Kashii T, et al. Multicentre prospective phase II trial of gefitinib for advanced non-small cell lung cancer with epidermal growth factor receptor mutations: results of the West Japan Thoracic Oncology Group trial (WJTOG0403). Br J Cancer 2008;98:907–914.
Lurje G, Lenz HJ . EGFR signaling and drug discovery. Oncology 2009;77:400–410.
Okabe T, Okamoto I, Tamura K, et al. Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res 2007;67:2046–2053.
Ciardiello F, Tortora G . A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res 2001;7:2958–2970.
Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 2011;29:2866–2874.
Han SW, Kim TY, Hwang PG, et al. Predictive and prognostic impact of epidermal growth factor receptor mutation in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol 2005;23:2493–2501.
Reck M . A major step towards individualized therapy of lung cancer with gefitinib: the IPASS trial and beyond. Expert Rev Anticancer Ther 2011;10:955–965.
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–957.
Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet 2008;372:1809–1818.
Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–742.
Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–128.
O'Byrne KJ, Gatzemeier U, Bondarenko I, et al. Molecular biomarkers in non-small-cell lung cancer: a retrospective analysis of data from the phase 3 FLEX study. Lancet Oncol 2011;12:795–805.
Herbst RS, Sun Y, Eberhardt WE, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol 2010;11:619–626.
Cappuzzo F, Ciuleanu T, Stelmakh L, et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol 2010;11:521–529.
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380–2388.
Reiter JL, Threadgill DW, Eley GD, et al. Comparative genomic sequence analysis and isolation of human and mouse alternative EGFR transcripts encoding truncated receptor isoforms. Genomics 2001;71:1–20.
Strausberg RL, Feingold EA, Grouse LH, et al. Generation and initial analysis of more than 15 000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA 2002;99:16899–16903.
Hynes NE, Lane HA . ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5:341–354.
Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006;366:2–16.
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039–1043.
Janku F, Stewart DJ, Kurzrock R . Targeted therapy in non-small-cell lung cancer-is it becoming a reality? Nat Rev Clin Oncol 2010;8:384.
Linardou H, Dahabreh IJ, Bafaloukos D, et al. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol 2009;6:352–366.
Yoon YK, Kim HP, Han SW, et al. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: implication for combinatorial approach. Mol Carcinog 2010;49:353–362.
Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–346.
Sordella R, Bell DW, Haber DA, et al. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004;305:1163–1167.
Roberts PJ, Stinchcombe TE, Der CJ, et al. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J Clin Oncol 2010;28:4769–4777.
Singh M, Lima A, Molina R, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol 2010;28:585–593.
Benesova L, Minarik M, Jancarikova D, et al. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res 2010;30:1667–1671.
Siena S, Sartore-Bianchi A, Di Nicolantonio F, et al. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst 2009;101:1308–1324.
Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005;2:e17.
Riely GJ, Politi KA, Miller VA, et al. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res 2006;12:7232–7241.
Zhou W, Ercan D, Janne PA, et al. Discovery of selective irreversible inhibitors for EGFR-T790M. Bioorg Med Chem Lett 2011;21:638–643.
Penzel R, Sers C, Chen Y, et al. EGFR mutation detection in NSCLC—assessment of diagnostic application and recommendations of the German Panel for Mutation Testing in NSCLC. Virchows Arch 2011;458:95–98.
Wu JY, Wu SG, Yang CH, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res 2008;14:4877–4882.
Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 2008;26:2442–2449.
Kumar A, Petri ET, Halmos B, et al. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol 2008;26:1742–1751.
Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006;12:6494–6501.
Gazdar AF . Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009;28 (Suppl 1):S24–S31.
Wang X, Wang M, MacLennan GT, et al. Evidence for common clonal origin of multifocal lung cancers. J Natl Cancer Inst 2009;101:560–570.
Weber F, Fukino K, Villalona-Calero M, et al. Limitations of single-strand conformation polymorphism analysis as a high-throughput method for the detection of EGFR mutations in the clinical setting. J Clin Oncol 2005;23:5847–5849.
Willmore-Payne C, Holden JA, Layfield LJ . Detection of epidermal growth factor receptor and human epidermal growth factor receptor 2 activating mutations in lung adenocarcinoma by high-resolution melting amplicon analysis: correlation with gene copy number, protein expression, and hormone receptor expression. Hum Pathol 2006;37:755–763.
Takano T, Ohe Y, Tsuta K, et al. Epidermal growth factor receptor mutation detection using high-resolution melting analysis predicts outcomes in patients with advanced non small cell lung cancer treated with gefitinib. Clin Cancer Res 2007;13:5385–5390.
Fukui T, Ohe Y, Tsuta K, et al. Prospective study of the accuracy of EGFR mutational analysis by high-resolution melting analysis in small samples obtained from patients with non-small cell lung cancer. Clin Cancer Res 2008;14:4751–4757.
Nomoto K, Tsuta K, Takano T, et al. Detection of EGFR mutations in archived cytologic specimens of non-small cell lung cancer using high-resolution melting analysis. Am J Clin Pathol 2006;126:608–615.
Hoshi K, Takakura H, Mitani Y, et al. Rapid detection of epidermal growth factor receptor mutations in lung cancer by the SMart-Amplification Process. Clin Cancer Res 2007;13:4974–4983.
Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1000 patients with lung adenocarcinoma: the NCI's Lung Cancer Mutation Consortium (LCMC). J Clin Oncol 2011;29: (Suppl, abstract CRA 7506).
Janne PA, Johnson BE . Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res 2006;12:4416s–4420s.
Ji H . Mechanistic insights into acquired drug resistance in epidermal growth factor receptor mutation-targeted lung cancer therapy. Cancer Sci 2010;101:1933–1938.
Shih JY, Gow CH, Yang PC . EGFR mutation conferring primary resistance to gefitinib in non-small-cell lung cancer. N Engl J Med 2005;353:207–208.
Toyooka S, Kiura K, Mitsudomi T . EGFR mutation and response of lung cancer to gefitinib. N Engl J Med 2005;352:2136.
Hammerman PS, Janne PA, Johnson BE . Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2009;15:7502–7509.
Jackman DM, Miller VA, Cioffredi LA, et al. Impact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non-small cell lung cancer patients: results of an online tumor registry of clinical trials. Clin Cancer Res 2009;15:5267–5273.
West H, Lilenbaum R, Harpole D, et al. Molecular analysis-based treatment strategies for the management of non-small cell lung cancer. J Thorac Oncol 2009;4:S1029–S1039; quiz S1041–S1042.
Rosell R, Taron M, Reguart N, et al. Epidermal growth factor receptor activation: how exon 19 and 21 mutations changed our understanding of the pathway. Clin Cancer Res 2006;12:7222–7231.
Pao W, Miller VA, Venkatraman E, et al. Predicting sensitivity of non-small-cell lung cancer to gefitinib: is there a role for P-Akt? J Natl Cancer Inst 2004;96:1117–1119.
Sugawa N, Ekstrand AJ, James CD, et al. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 1990;87:8602–8606.
Pedersen MW, Meltorn M, Damstrup L, et al. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann Oncol 2001;12:745–760.
Antonyak MA, Moscatello DK, Wong AJ . Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J Biol Chem 1998;273:2817–2822.
Chu CT, Everiss KD, Wikstrand CJ, et al. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem J 1997;324:855–861.
Huang HS, Nagane M, Klingbeil CK, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997;272:2927–2935.
Frederick L, Wang XY, Eley G, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000;60:1383–1387.
Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005;353:2012–2024.
Li B, Yuan M, Kim IA, et al. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 2004;23:4594–4602.
Sasaki H, Kawano O, Endo K, et al. EGFRvIII mutation in lung cancer correlates with increased EGFR copy number. Oncol Rep 2007;17:319–323.
Ji H, Zhao X, Yuza Y, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci USA 2006;103:7817–7822.
Ohtsuka K, Ohnishi H, Fujiwara M, et al. Abnormalities of epidermal growth factor receptor in lung squamous-cell carcinomas, adenosquamous carcinomas, and large-cell carcinomas: tyrosine kinase domain mutations are not rare in tumors with an adenocarcinoma component. Cancer 2007;109:741–750.
Li D, Ji H, Zaghlul S, et al. Therapeutic anti-EGFR antibody 806 generates responses in murine de novo EGFR mutant-dependent lung carcinomas. J Clin Invest 2007;117:346–352.
Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med 2005;353:133–144.
Hirsch FR, Varella-Garcia M, Bunn Jr PA, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 2003;21:3798–3807.
Awaya H, Takeshima Y, Furonaka O, et al. Gene amplification and protein expression of EGFR and HER2 by chromogenic in situ hybridisation and immunohistochemistry in atypical adenomatous hyperplasia and adenocarcinoma of the lung. J Clin Pathol 2005;58:1076–1080.
Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 2005;6:279–286.
Hoff K, Jorgensen JT, Muller S, et al. Visualization of FISH Probes by dual-color chromogenic in situ hybridization. Am J Clin Pathol 2010;133:205–211.
Scagliotti GV, Selvaggi G, Novello S, et al. The biology of epidermal growth factor receptor in lung cancer. Clin Cancer Res 2004;10:4227s–4232s.
Khambata-Ford S, Harbison CT, Hart LL, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol 2010;28:918–927.
Scartozzi M, Bearzi I, Berardi R, et al. Epidermal growth factor receptor (EGFR) status in primary colorectal tumors does not correlate with EGFR expression in related metastatic sites: implications for treatment with EGFR-targeted monoclonal antibodies. J Clin Oncol 2004;22:4772–4778.
Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005;23:1803–1810.
Wheeler DL, Dunn EF, Harari PM . Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol 2010;7:493–507.
Li AR, Chitale D, Riely GJ, et al. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J Mol Diagn 2008;10:242–248.
Endoh H, Ishibashi Y, Yamaki E, et al. Immunohistochemical analysis of phosphorylated epidermal growth factor receptor might provide a surrogate marker of EGFR mutation. Lung Cancer 2009;63:241–246.
Hijiya N, Miyawaki M, Kawahara K, et al. Phosphorylation status of epidermal growth factor receptor is closely associated with responsiveness to gefitinib in pulmonary adenocarcinoma. Hum Pathol 2008;39:316–323.
Yu J, Kane S, Wu J, et al. Mutation-specific antibodies for the detection of EGFR mutations in non-small-cell lung cancer. Clin Cancer Res 2009;15:3023–3028.
Brevet M, Arcila M, Ladanyi M . Assessment of EGFR mutation status in lung adenocarcinoma by immunohistochemistry using antibodies specific to the two major forms of mutant EGFR. J Mol Diagn 2010;12:169–176.
Zhang Z, Stiegler AL, Boggon TJ, et al. EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget 2010;1:497–514.
Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554.
Vakiani E, Solit DB . KRAS and BRAF: drug targets and predictive biomarkers. J Pathol 2011;223:219–229.
Sun L, Zhang Q, Luan H, et al. Comparison of KRAS and EGFR gene status between primary non-small cell lung cancer and local lymph node metastases: implications for clinical practice. J Exp Clin Cancer Res 2011;30:30.
Paez D, Pare L, Espinosa I, et al. Immunoglobulin G fragment C receptor polymorphisms and KRAS mutations: are they useful biomarkers of clinical outcome in advanced colorectal cancer treated with anti-EGFR-based therapy? Cancer Sci 2011;101:2048–2053.
Mao C, Qiu LX, Liao RY, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer 2011;69:272–278.
Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010;17:77–88.
Toschi L, Cappuzzo F . Clinical implications of MET gene copy number in lung cancer. Future Oncol 2011;6:239–247.
Suda K, Murakami I, Katayama T, et al. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res 2011;16:5489–5498.
McDermott U, Pusapati RV, Christensen JG, et al. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res 2011;70:1625–1634.
Tiseo M, Gelsomino F, Boggiani D, et al. EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer 2011;71:241–243.
Murakami S, Yokose T, Saito H, et al. Recurrent EML4-ALK-associated lung adenocarcinoma with a slow clinical course. Lung Cancer 2011;69:361–364.
Normant E, Paez G, West KA, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2010;30:2581–2586.
Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734–1739.
Barton S, Starling N, Swanton C . Predictive molecular markers of response to epidermal growth factor receptor (EGFR) family-targeted therapies. Curr Cancer Drug Targets 2010;10:799–812.
van Eijk R, Licht J, Schrumpf M, et al. Rapid KRAS, EGFR, BRAF and PIK3CA mutation analysis of fine needle aspirates from non-small-cell lung cancer using allele-specific qPCR. PLoS One 2011;6:e17791.
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753–762.
Schmid K, Oehl N, Wrba F, et al. EGFR/KRAS/BRAF mutations in primary lung adenocarcinomas and corresponding locoregional lymph node metastases. Clin Cancer Res 2009;15:4554–4560.
Nguyen KS, Kobayashi S, Costa DB . Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 2009;10:281–289.
Morgillo F, Kim WY, Kim ES, et al. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res 2007;13:2795–2803.
Miyamae Y, Shimizu K, Hirato J, et al. Significance of epidermal growth factor receptor gene mutations in squamous cell lung carcinoma. Oncol Rep 2011;25:921–928.
Tochigi N, Dacic S, Nikiforova M, et al. Adenosquamous carcinoma of the lung: a microdissection study of KRAS and EGFR mutational and amplification status in a western patient population. Am J Clin Pathol 2011;135:783–789.
Ladanyi M, Pao W . Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol 2008;21 (Suppl 2):S16–S22.
Chen C, Jiang XL, Zhang CC, et al. Correlation of genes associated with drug response to prognosis of large cell lung carcinoma. Chin J Cancer 2011;30:497–504.
De Pas TM, Giovannini M, Manzotti M, et al. Large-cell neuroendocrine carcinoma of the lung harboring EGFR mutation and responding to Gefitinib. J Clin Oncol 2011;29:e819–e822.
Jiang X, Liu Y, Chen C, et al. The value of biomarkers in patients with sarcomatoid carcinoma of the lung: molecular analysis of 33 cases. Clin Lung Cancer 2012 (in press).
Hammerman P, Sos ML, Ramos A, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov 2011;1:78–89.
Travis WD, Brambilla E, Noguchi M, et al. International association for the study of lung cancer/American Thoracic Society/European Respiratory Society: international multidisciplinary classification of lung adenocarcinoma: executive summary. Proc Am Thorac Soc 2011;8:381–385.
Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994;263:1281–1284.
Palmer RH, Vernersson E, Grabbe C, et al. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 2009;420:345–361.
Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008;8:11–23.
Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol 2009;174:661–670.
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–4253.
Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol 2009;22:508–515.
Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol 2008;3:13–17.
Shaw AT, Solomon B . Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res 2011;17:2081–2086.
Horn L, Sandler A . Epidermal growth factor receptor inhibitors and antiangiogenic agents for the treatment of non-small cell lung cancer. Clin Cancer Res 2009;15:5040–5048.
Horn L, Pao W . EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol 2009;27:4232–4235.
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 2009;15:3143–3149.
Choi YL, Takeuchi K, Soda M, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res 2008;68:4971–4976.
Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res 2008;14:6618–6624.
Polgar D, Leisser C, Maier S, et al. Truncated ALK derived from chromosomal translocation t(2;5)(p23;q35) binds to the SH3 domain of p85-PI3K. Mutat Res 2005;570:9–15.
Garber K . ALK, lung cancer, and personalized therapy: portent of the future? J Natl Cancer Inst 2010;102:672–675.
Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009;115:1723–1733.
Zhang X, Zhang S, Yang X, et al. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol Cancer 2010;9:188.
Sasaki T, Rodig SJ, Chirieac LR, et al. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010;46:1773–1780.
Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res 2009;15:5216–5223.
McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res 2008;68:3389–3395.
Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011;71:6051–6060.
Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci USA 2011;108:7535–7540.
Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 2011;12:1004–1012.
Just PA, Cazes A, Audebourg A, et al. Histologic subtypes, immunohistochemistry, FISH or molecular screening for the accurate diagnosis of ALK-rearrangement in lung cancer: a comprehensive study of Caucasian non-smokers. Lung Cancer 2012 (in press).
McLeer-Florin A, Moro-Sibilot D, Melis A, et al. Dual IHC and FISH testing for ALK gene rearrangement in lung adenocarcinomas in a routine practice: A French Study. J Thorac Oncol 2012 (in press).
Jorissen RN, Walker F, Pouliot N, et al. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 2003;284:31–53.
Nicholas MK, Lukas RV, Jafri NF, et al. Epidermal growth factor receptor - mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res 2006;12:7261–7270.
Yang XD, Jia XC, Corvalan JR, et al. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res 1999;59:1236–1243.
Kawamoto T, Sato JD, Le A, et al. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc Natl Acad Sci USA 1983;80:1337–1341.
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004;351:337–345.
Hinoda Y, Sasaki S, Ishida T, et al. Monoclonal antibodies as effective therapeutic agents for solid tumors. Cancer Sci 2004;95:621–625.
Baselga J, Arteaga CL . Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol 2005;23:2445–2459.
Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003;290:2149–2158.
Tam IY, Leung EL, Tin VP, et al. Double EGFR mutants containing rare EGFR mutant types show reduced in vitro response to gefitinib compared with common activating missense mutations. Mol Cancer Ther 2009;8:2142–2151.
Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 2008;359:366–377.
Bell DW, Gore I, Okimoto RA, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 2005;37:1315–1316.
Ercan D, Zejnullahu K, Yonesaka K, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010;29:2346–2356.
Hopper-Borge EA, Nasto RE, Ratushny V, et al. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin Ther Targets 2009;13:339–362.
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786–792.
Eberhard DA, Giaccone G, Johnson BE . Biomarkers of response to epidermal growth factor receptor inhibitors in Non-Small-Cell Lung Cancer Working Group: standardization for use in the clinical trial setting. J Clin Oncol 2008;26:983–994.
Okamoto K, Okamoto I, Okamoto W, et al. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res 2011;70:10402–10410.
Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–1622.
Engelman JA, Janne PA . Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008;14:2895–2899.
Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 2010;28:357–360.
Govindan R . A review of epidermal growth factor receptor/HER2 inhibitors in the treatment of patients with non-small-cell lung cancer. Clin Lung Cancer 2010;11:8–12.
Ogino S, Meyerhardt JA, Irahara N, et al. KRAS mutation in stage III colon cancer and clinical outcome following intergroup trial CALGB 89803. Clin Cancer Res 2009;15:7322–7329.
Mink SR, Vashistha S, Zhang W, et al. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol Cancer Res 2010;8:809–820.
Wang W, Li Q, Yamada T, et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res 2009;15:6630–6638.
Lemos C, Jansen G, Peters GJ . Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors. Br J Cancer 2008;98:857–862.
Inoue A, Suzuki T, Fukuhara T, et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol 2006;24:3340–3346.
Krumbach R, Schuler J, Hofmann M, et al. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer 2011;47:1231–1243.
Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2005;2:e313.
Tam IY, Chung LP, Suen WS, et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res 2006;12:1647–1653.
Kawano O, Sasaki H, Endo K, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 2006;54:209–215.
Sos ML, Koker M, Weir BA, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009;69:3256–3261.
Vivanco I, Rohle D, Versele M, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci USA 2010;107:6459–6464.
Yamamoto C, Basaki Y, Kawahara A, et al. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res 2010;70:8715–8725.
Kohno T, Takahashi M, Manda R, et al. Inactivation of the PTEN/MMAC1/TEP1 gene in human lung cancers. Genes Chromosomes Cancer 1998;22:152–156.
Soria JC, Lee HY, Lee JI, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res 2002;8:1178–1184.
Ellis LM, Hicklin DJ . Resistance to targeted therapies: refining anticancer therapy in the Era of molecular oncology. Clin Cancer Res 2009;15:7471–7478.
Harris TJ, McCormick F . The molecular pathology of cancer. Nat Rev Clin Oncol 2010;7:251–265.
Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet 2007;39:347–351.
Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455:1069–1075.
Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 2008;9:962–972.
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73.
Santis G, Angell R, Nickless G, et al. Screening for EGFR and KRAS mutations in endobronchial ultrasound derived transbronchial needle aspirates in non-small cell lung cancer using COLD-PCR. PLoS One 2010;6:e25191.
Reinersman JM, Johnson ML, Riely GJ, et al. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol 2011;6:28–31.
Shigematsu H, Gazdar AF . Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer 2006;118:257–262.
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–954.
Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–2051.
Leicht DT, Balan V, Kaplun A, et al. Raf kinases: function, regulation and role in human cancer. Biochem Biophys Acta 2007;1773:1196–1212.
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008;26:5705–5712.
Yoon J, Koo KH, Choi KY . MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res 2011;71:445–453.
Yousem SA, Nikiforova M, Nikiforov Y . The histopathology of BRAF-V600E-mutated lung adenocarcinoma. Am J Surg Pathol 2008;32:1317–1321.
De Oliveira Duarte Achcar R, Nikiforova MN, Yousem SA . Micropapillary lung adenocarcinoma: EGFR, K-ras, and BRAF mutational profile. Am J Clin Pathol 2009;131:694–700.
Qi J, McTigue MA, Rogers A, et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res 2011;71:1081–1091.
Yarden Y, Sliwkowski MX . Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–137.
Bianco R, Shin I, Ritter CA, et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene 2003;22:2812–2822.
She QB, Solit D, Basso A, et al. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res 2003;9:4340–4346.
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–1765.
Herbst RS, Heymach JV, Lippman SM . Lung cancer. N Engl J Med 2008;359:1367–1380.
Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest 2006;116:2695–2706.
Yatabe Y, Matsuo K, Mitsudomi T . Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J Clin Oncol 2010;29:2972–2977.
Zhou Q, Zhang XC, Chen ZH, et al. Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non-small-cell lung cancer. J Clin Oncol 2011;29:3316–3321.
Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141:69–80.
Costa DB, Nguyen KS, Cho BC, et al. Effects of erlotinib in EGFR mutated non-small cell lung cancers with resistance to gefitinib. Clin Cancer Res 2008;14:7060–7067.
Seki T, Hagiya M, Shimonishi M, et al. Organization of the human hepatocyte growth factor-encoding gene. Gene 1991;102:213–219.
Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009;27:1667–1674.
Beau-Faller M, Ruppert AM, Voegeli AC, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol 2008;3:331–339.
Longati P, Bardelli A, Ponzetto C, et al. Tyrosines1234-1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994;9:49–57.
Eder JP, Vande Woude GF, Boerner SA, et al. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res 2009;15:2207–2214.
Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med 2006;355:983–991.
Simon GR, Sharma S, Cantor A, et al. ERCC1 expression is a predictor of survival in resected patients with non-small cell lung cancer. Chest 2005;127:978–983.
Gandara DR, Grimminger P, Mack PC, et al. Association of epidermal growth factor receptor activating mutations with low ERCC1 gene expression in non-small cell lung cancer. J Thorac Oncol 2010;5:1933–1938.
Lee KH, Min HS, Han SW, et al. ERCC1 expression by immunohistochemistry and EGFR mutations in resected non-small cell lung cancer. Lung Cancer 2008;60:401–407.
Joerger M, deJong D, Burylo A, et al. Tubuline, BRCA1, ERCC1, Abraxas, RAP80 mRNA expression, p53/p21 immunohistochemistry and clinical outcome in patients with advanced non small-cell lung cancer receiving first-line platinum-gemcitabine chemotherapy. Lung Cancer 2011;74:310–317.
Gomez-Roca C, Raynaud CM, Penault-Llorca F, et al. Differential expression of biomarkers in primary non-small cell lung cancer and metastatic sites. J Thorac Oncol 2009;4:1212–1220.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123–132.
Yang CH, Yu CJ, Shih JY, et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J Clin Oncol 2008;26:2745–2753.
Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005;102:7665–7670.
Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011;17:1169–1180.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Cheng, L., Alexander, R., MacLennan, G. et al. Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol 25, 347–369 (2012). https://doi.org/10.1038/modpathol.2011.215
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.215
Keywords
This article is cited by
-
A-to-I edited miR-411-5p targets MET and promotes TKI response in NSCLC-resistant cells
Oncogene (2023)
-
Leukotriene B4 receptor-2 contributes to KRAS-driven lung tumor formation by promoting interleukin-6-mediated inflammation
Experimental & Molecular Medicine (2021)
-
Integrative analysis of oncogenic fusion genes and their functional impact in colorectal cancer
British Journal of Cancer (2018)
-
Decrease of FSTL1-BMP4-Smad signaling predicts poor prognosis in lung adenocarcinoma but not in squamous cell carcinoma
Scientific Reports (2017)
-
Molecular genetic studies on EGFR, KRAS, BRAF, ALK, PIK3CA, PDGFRA, and DDR2 in primary pulmonary adenoid cystic carcinoma
Diagnostic Pathology (2015)
