
Abstract
Although DNA methylation profiles in breast cancer have been connected to breast cancer molecular subtype, there have been no studies of the association of DNA methylation with stem cell phenotype. This study was designed to evaluate the promoter CpG island methylation of 15 genes in relation to breast cancer subtype, and to investigate whether the patterns of CpG island methylation in each subtype are associated with their cancer stem cell phenotype represented by CD44+/CD24− and ALDH1 expression. We performed MethyLight analysis of the methylation status of 15 promoter CpG island loci involved in breast cancer progression (APC, DLEC1, GRIN2B, GSTP1, HOXA1, HOXA10, IGF2, MT1G, RARB, RASSF1A, RUNX3, SCGB3A1, SFRP1, SFRP4, and TMEFF2) and determined cancer stem cell phenotype by CD44/CD24 and ALDH1 immunohistochemistry in 36 luminal A, 33 luminal B, 30 luminal–HER2, 40 HER2 enriched, and 40 basal-like subtypes of breast cancer. The number of CpG island loci methylated differed significantly between subtypes, and was highest in the luminal–HER2 subtype and lowest in the basal-like subtype. Methylation frequencies and levels in 12 of the 15 genes differed significantly between subtypes, and the basal-like subtype had significantly lower methylation frequencies and levels in nine of the genes than the other subtypes. CD44+/CD24− and ALDH1+ putative stem cell populations were most enriched in the basal-like subtype. Methylation of promoter CpG islands was significantly lower in CD44+/CD24-cell (+) tumors than in CD44+/CD24-cell (−) tumors, even within the basal-like subtype. ALDH1 (+) tumors were also less methylated than ALDH1 (−) tumors. Our findings showed that promoter CpG island methylation was different in relation to breast cancer subtype and stem cell phenotype of tumor, suggesting that breast cancers have distinct patterns of CpG island methylation according to molecular subtypes and these are associated with different stem cell phenotypes of the tumor.
Similar content being viewed by others


Main
Breast cancer is a heterogeneous disease with different biological characteristics and clinical behaviors. Five major molecular subtypes (luminal A, luminal B, HER2+, basal-like, and normal breast-like) have been identified by comprehensive gene expression profiling and are associated with different clinical outcomes.1, 2, 3 Luminal A breast cancers express hormone receptor, have favorable outcomes, and can be treated by hormone therapy, while basal-like and HER2+ breast cancers do not express hormone receptors and have poor prognoses. Although HER2+ breast cancers can be treated by targeted therapy using trastuzumab or lapatinib, no specific therapy is available for the basal-like subtype.4, 5
As in other types of human cancer, breast cancer development involves the accumulation of both genetic alterations such as amplification of oncogenes and mutation or loss of tumor suppressor genes (TSGs) and epigenetic alterations such as DNA methylation and histone modification.6, 7 Cancer-related DNA methylation involves genomic DNA hypomethylation associated with chromosomal instability and hypermethylation of promoter CpG islands, the latter representing an alternative mechanism for inactivating TSGs, resulting in their transcriptional silencing.8, 9 In breast cancer, promoter CpG island hypermethylation has been described for genes involved in all aspects of cellular function.7 There are reports that promoter CpG island hypermethylation is associated with various histopathologic characteristics of breast cancers, including histologic type,10, 11 tumor grade,12, 13 hormone receptor, and HER2/neu status.14, 15, 16, 17 Array-based comprehensive DNA methylation profiling has shown that breast cancer molecular subtypes have their own methylation profiles.18, 19, 20, 21 Moreover, these different methylation profiles were evident throughout the CpG islands of the genome, not limited to functional genes.20 Kamalakaran et al20 reported that the methylation patterns of differentially methylated genes in luminal A tumors were similar to those identified in CD24+ luminal epithelial cells, and those in basal-like tumors resembled those of CD44+ breast progenitor cells, suggesting that the methylation patterns of the breast cancer subtypes reflect the methylation patterns of their cells of origin.
The idea that the different breast cancer subtypes derive from different cell types would mean that basal-like tumors derive from bi-potential mammary stem cells and luminal tumors from luminal progenitor cells,22 although recent studies suggest that the luminal progenitor population is a target for transformation in BRCA1-associated basal tumors.23, 24 In breast cancer, lin−/CD44+/CD24−/low cells have been proposed as candidate breast cancer stem cells based on xenotransplant assays in NOD/SCID mice.25 Aldehyde dehydrogenase (ALDH) activity has also been suggested as a putative marker of normal and malignant mammary stem cells.26 In a previous study,27 we investigated the expression of 12 proteins including CD44, CD24, and ALDH1, which were differentially expressed in breast cancer cells with more differentiated luminal (CD24+) and stem cell-like characteristics (CD44+),28 in different breast cancer subtypes and found that CD44+/CD24− and ALDH1+ cancer cells were enriched in basal-like breast cancers.
In this study, we evaluated promoter CpG island methylation in various breast cancer subtypes using 15 genes (APC, DLEC1, GRIN2B, GSTP1, HOXA1, HOXA10, IGF2, MT1G, RARB, RASSF1A, RUNX3, SCGB3A1, SFRP1, SFRP4, and TMEFF2) previously implicated in breast cancer progression.13 We investigated in particular whether the distinct patterns of CpG island methylation in each subtype reflected their cancer stem cell phenotypes as represented by CD44+/CD24− phenotype and ALDH1 expression.
Materials and methods
Patients and Tissue Specimens
Formalin-fixed, paraffin-embedded archival tissue from 129 patients who underwent surgical resection for invasive breast cancer between 2004 and 2009 were selected from the files of the Department of Pathology, Seoul National University Bundang Hospital, based on immunohistochemical findings of ER, PR, HER2, Ki-67, cytokeratin 5/6 and EGFR, and breast cancer subtype defined by immunohistochemical profile. In addition, we included immunohistochemical and methylation data for 50 cases of invasive breast cancer from a previous study.13 All patients were female, with a mean age of 51 (range, 20–85 years). Clinicopathologic information was obtained by reviewing pathology reports and hematoxylin and eosin-stained sections. The following histopathologic variables were determined: histologic subtype, tumor size, T stage, N stage, Nottingham combined histologic grade, lymphovascular invasion, tumor border, and presence or absence of ducal carcinoma in situ component. All cases were independently reviewed by two breast pathologists (SYP and HEL). The study was approved by the institutional review board of Seoul National University Bundang Hospital (protocol # B-1005-100-302).
MethyLight Analysis
Three to five serial sections (4-μm thick) from each representative paraffin block were stained with hematoxylin and eosin. Tumor areas with >70% neoplastic cells were marked and manually dissected under a microscope. Dissected tissues were subjected to tissue lysis using proteinase K lysis buffer at 55 °C for 24–48 h. Bisulfite modification of the digested samples was performed using an EZ DNA methylation kit (Zymo Research, Orange, CA, USA). DNA methylation analyses were performed with the MethyLight assay. MethyLight reaction primers and probes for the 15-CpG island loci (APC, DLEC1, GRIN2B, GSTP1, HOXA1, HOXA10, IGF2, MT1G, RARB, RASSF1A, RUNX3, SCGB3A1, SFRP1, SFRP4, and TMEFF20) were used as described previously.13 Briefly, MethyLight PCR was performed in 30-μl volumes containing 0.2 mM deoxyribonucleotide triphosphates, 0.3 μM forward and reverse primers, 0.05 μM probe, 3.5 mM MgCl2, 0.05% gelatin, 0.01% Tween-20, 1 × PCR buffer, and 0.5 U of AmpliTaq Gold polymerase. The amplification conditions for all primers consisted of 95 °C for 10 min, followed by 50 cycles of 95 °C for 20 s, and 59 °C for 40 s. ALU repeats were used as an internal reference to normalize input DNA and to generate a standard curve. Bisulfite-modified placental genomic DNA treated with M.SssI was used as a control reaction for the ALU repeat and selected promoter CpG loci. The percentage of methylated reference (PMR) for each genetic locus was calculated as 100 times the GENE/ALU methylation ratio of each sample divided by the GENE/ALU methylation ratio of reference DNA treated with M.SssI. The methylation status of each sample was considered positive when PMR >4, a cutoff value based on validated data.29
Immunohistochemical Analyses and Scoring
In all, 4-μm thick tissue sections were cut, dried, deparaffinized, and rehydrated following the standard procedures. All the sections were subjected to heat-induced antigen retrieval. Immunohistochemical staining was carried out in a BenchMark XT autostainer (Ventana Medical Systems, Tucson, AZ, USA) using an i-View detection kit (Ventana Medical Systems) for ER (1:100; clone SP1; Labvision), PR (1:70; PgR 636; Dako), HER2 (1:700; polyclonal; Dako), p53 (1:600; D07; Dako), Ki-67 (1:250; MIB-1; Dako), cytokeratin 5/6 (1:50; clone D5/16 B4; Dako), epidermal growth factor receptor (EGFR) (EGFR pharmDx™, Dako), CD44 (1:200; Clone 156-3C11; Neomarkers), CD24 (1:100; Clone SN3b; Neomarkers), and ALDH1 (1:100; Clone 44; BD Biosciences). Double immunostaining with antibodies to CD44 and CD24 was performed using the EnVision G∣2 Doublestain System (Dako). CD44 was detected with diaminobenzidene and CD24 was detected using Permanent Red. We confirmed the accuracy of double immunostaining by comparing it with single immunostaining for CD44 and CD24, separately.
ER and PR were regarded as positive if there were at least 1% positive tumor nuclei, according to the ASCO/CAP guidelines.30 Expression of HER2 and EGFR was scored as follows: 0, no staining; 1+, weak and incomplete membranous staining in ≥10% of the tumor cells; 2+, weak to moderate, complete membranous staining in ≥10% of the tumor cells; 3+, strong, complete membranous staining in ≥30% of the tumor cells. Any positive staining was regarded as positive for EGFR, and 3+ was considered positive for HER2. For cytokeratin 5/6, cases with any positive membranous staining were grouped as positive. For p53, cases with ≥10% positive staining were grouped as positive. The proportions of ALDH1 positive and CD44+/CD24− tumor cells were counted semiquantitatively and any positive cells, and ≥10% positive cells were grouped as positive, respectively.
Fluorescence In Situ Hybridization Assays for HER2 Gene Amplification
To determine HER2 gene amplification, we used the PathVysion (Vysis, Downers Grove, IL, USA) assay and evaluated it as previously described.31 At least 50 cells were examined for each case, and the HER2 gene was considered to be amplified in tumors with ratios of HER-2 to CEP17 ≥2.2.
Definition of Breast Tumor Subtypes
Breast cancer subtypes were defined according to Voduc et al32 and were categorized as follows: luminal A (ER+ or PR+, HER2–, Ki-67 <14%) luminal B (ER+ or PR+, HER2–, Ki-67 ≥14%), luminal–HER2 (ER+ or PR+, HER2+), HER2 enriched (ER–, PR–, HER2+) and basal-like (ER–, PR–, HER2–, cytokeratin 5/6+ or EGFR+). HER2 positivity was determined based on FISH (fluorescence in situ hybridization) results as described. The cases consisted of 36 luminal A, 33 luminal B, 30 luminal–HER2, 40 HER2 enriched, and 40 basal-like subtypes.
Statistical Analysis
Statistical analysis was performed with SPSS software (version 15.0, SPSS, Chicago, IL, USA). The χ2 test or Fisher’s exact test was used when comparing categorical variables. Differences between continuous data including PMR values for tumor subtypes were analyzed by ANOVA or the Kruskal–Wallis test, as appropriate. If differences between the two independent groups were involved, the independent-samples t-test or Mann–Whitney test was used. P-values <0.05 were considered statistically significant. All P-values reported were two-sided.
Results
Clinicopathologic Characteristics of Tumors
Patient and tumor characteristics in relation to subtype are summarized in Table 1. Significant differences were found for the age of patients (P=0.017) and multiple comparison indicated that the patients with basal-like cancers were significantly younger than those with HER2 enriched subtype (P=0.030). Histologic grade, p53 overexpression, and Ki-67 proliferation index were significantly different between the subtypes (P<0.001). The basal-like and HER2 enriched subtypes had high histologic grades and a higher frequency of p53 overexpression than the other subtypes. Ki-67 proliferation indices were significantly higher in the basal-like, HER2 enriched and luminal B subtypes.
CpG Island Methylation According to Breast Cancer Subtype
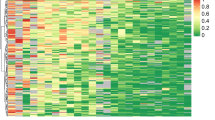
Using MethyLight analysis, we examined the promoter CpG island methylation of a panel of 15 genes shown to be involved in breast cancer progression in a previous study.13 The different samples varied in levels of methylation of the 15 genes, as shown in the methylation map of the PMR values obtained for each CpG island locus (Figure 1). First, we compared the number of methylated CpG islands according to subtype. The number of CpG island loci methylated (PMR>4) varied from 1 to 13 in the luminal A and luminal B subtypes, from 5 to 14 in the luminal–HER2 subtype, from 1 to 12 in the HER2 enriched subtype, and from 0 to 7 in the basal-like subtype (Figure 2). The number of CpG island loci methylated was significantly different between subtypes (Kruskal–Wallis test; P<0.001). It was highest in the luminal–HER2 subtype, showing significant difference with luminal A, HER2 enriched, and basal-like subtypes (luminal–HER2 vs luminal A, P<0.001; luminal–HER2 vs HER2 enriched, P=0.022; luminal–HER2 vs basal-like, P<0.001) and lowest in basal-like subtype with significant differences with the other subtypes (P<0.001). There were no differences between the luminal–HER2 and luminal B subtypes, and between the HER2 enriched and luminal subtypes.

Methylation map of the PMR values obtained for each CpG island locus in the luminal A, luminal B, luminal–HER2, HER2 enriched, and basal-like subtypes of breast cancer. Colored boxes represent four classes of methylation level [0<PMR<4 (light blue), 4≤PMR<20 (light orange), 20≤PMR <50 (orange), PMR≥50 (brown)]. Hypomethylation of the basal-like subtype is apparent.

Box plot depicting the number of methylated genes among the 15-CpG island loci in the subtypes of breast cancer. The box shows the 25th–75th percentile, the horizontal line inside the box represents the median, the whiskers extend to the 10th and 90th percentiles, and the outlying black circles are individual data points outside the 10th and 90th percentiles. The number of CpG island loci methylated is highest in the luminal–HER2 subtype and lowest in the basal-like subtype.
We also compared methylation frequencies and levels for each locus (Tables 2 and 3). Significant differences in methylation frequencies and levels were found between the subtypes for 12 genes (APC, DLEC1, GRIN2B, GSTP1, HOXA1, MT1G, RARB, RASSF1A, RUNX3, SCGB3A1, SFRP1, and TMEFF2) but not for HOXA10, IGF2, and SFRP4. Specifically, the basal-like subtype had significantly lower methylation frequencies and levels in those 12 genes than the luminal–HER and HER enriched subtypes, lower methylation frequencies and levels in 10 genes (APC, DLEC1, GRIN2B, GSTP1, HOXA1, RASSF1A, RUNX3, SCGB3A1, SFRP1, and TMEFF2) than the luminal B subtype, and lower methylation frequencies and levels in nine genes (APC, DLEC1, GSTP1, HOXA1, RASSF1A, RUNX3, SCGB3A1, SFRP1, and TMEFF2) than the luminal A subtype.
When the luminal–HER2 and HER2 enriched subtypes were compared, the luminal–HER2 subtype had significantly higher methylation frequencies and levels for DLEC1 (P=0.027, P=0.015, respectively), GSTP1 (P=0.043, P=0.002, respectively), RUNX3 (P=0.018, P=0.002, respectively), SCGB3A1 (P=0.007, P=0.002, respectively), and SFRP1 (P=0.022, P=0.011, respectively). The luminal–HER2 and luminal B subtypes differed in methylation frequency and level only for SCGB3A1 (P=0.008, P=0.018, respectively). The luminal B and luminal A subtypes differed in methylation frequency for HOXA1 (P=0.032) and RARB (P=0.048), and in methylation levels for APC (P=0.041), HOXA1 (P=0.006), and SFRP1 (P=0.026).
When ER-positive and ER-negative groups were analyzed collectively, APC, DLEC1, HOXA1, RASSF1A, RUNX3, SCGB3A1, and SFRP1 had significantly higher methylation frequencies and levels in the ER-positive groups. GSTP1 and TMEFF2 were more highly methylated in the ER-positive group, but only in terms of the methylation frequency (Figure 3a and b).

Comparison of methylation frequencies (a) and levels (b) of CpG island loci between ER-negative and ER-positive breast cancers. Methylation levels are given as means and SE (error bar). (a) Methylation frequencies of APC, DLEC1, GSTP1, HOXA1, RASSF1A, RUNX3, SCGB3A1, SFRP1, and TMEFF2 are significantly higher in ER-positive than in ER-negative tumors. (b) Methylation levels are significantly higher in ER-positive than in ER-negative tumors for APC, DLEC1, HOXA1, RASSF1A, RUNX3, SCGB3A1, and SFRP1, but not for GSTP1 and TMEFF2.
Association of CpG Island Methylation with the Expression of Stem Cell Markers
We have shown above that promoter CpG islands are less methylated in the basal-like subtypes than the other subtypes. To see whether this finding was associated with the stem cell phenotype of the tumors, we examined stem cell markers in 171 cases by immunohistochemistry. CD44+/CD24− putative stem cell populations differed between subtypes, being most enriched in the basal-like subtype (P=0.009 by the Kruskal–Wallis test). The frequency of CD44+/CD24-cell (+) tumors was significantly higher in the basal-like subtype than the other subtypes (luminal A vs basal-like, P=0.010; luminal B vs basal-like, P=0.011; luminal–HER2 vs basal-like, P<0.001; HER2 enriched vs basal-like, P=0.001 by χ2 test; Figures 4a and 5). ALDH1 (+) cell populations also differed between subtypes (P=0.001 by the Kruskal–Wallis test), being again most enriched in the basal-like subtype. ALDH1 positivity was higher in basal-like subtype, being significantly different from the luminal A, luminal B, and luminal–HER2 subtypes (P<0.001, P=0.002, P=0.029 by the χ2 test, respectively; Figures 4b and 5). When CD44+/CD24-cell (+) tumors were compared with CD44+/CD24-cell (−) tumors, the number of methylated genes was significantly lower in the CD44+/CD24-cell (+) tumors (P<0.001; Figure 6a). Methylation frequencies and levels of APC, DLEC1, GRIN2B, GSTP1, HOXA1, MT1G, RASSF1A, and TMEFF2 were significantly lower in the CD44+/CD24-cell (+) group than the CD44+/CD24-cell (−) group (Table 4). ALDH1-positive group also had significantly lower number of methylated genes than the ALDH1-negative group (P=0.023; Figure 6b). Compared with ALDH1-negative group, ALDH1-positive group had significantly lower methylation frequencies and levels for APC (48 vs 28%, P=0.047; 1.46 vs 0, median, P=0.013), DLEC1 (44 vs 22%, P=0.022; 2.27 vs 0, median, P=0.001), and RASSF1A (77 vs 50%, P=0.002; 15.32 vs 3.62, median, P=0.004). In the subgroup analyses of basal-like and non-basal-like subtypes, the number of methylated genes remained lower in the CD44+/CD24-cell (+) group of basal-like subtype than in the CD44+/CD24-cell (−) group (P=0.021; Figure 6c) and tended to be low in CD44+/CD24-cell (+) group of non-basal-like subtypes (P=0.093; Figure 6d). However, the subgroup analyses revealed no differences with respect to ALDH1.

Comparison of frequencies of CD44+/CD24-cell (+) tumors (a) and ALDH1 (+) tumors (b) in the luminal A, luminal B, luminal–HER2, HER2 enriched, and basal-like subtypes of breast cancer. CD44+/CD24-cell (+) and ALDH1 (+) tumors are most frequently found in the basal-like subtype.

Representative examples of CD44 (brown)/CD24 (red) and ALDH1 expression in the luminal B, HER2 enriched, and basal-like subtypes of breast cancer. While the luminal B and HER2 enriched subtypes are mainly composed of CD44−/CD24+ cells, the basal-like subtype contains predominantly CD44+/CD24− cells. ALDH1 is expressed in the HER2 enriched and basal-like subtypes, not in the luminal B subtype.

Comparison of numbers of methylated genes in relation to CD44+/CD24− (a) and ALDH1 expression (b). The number of methylated genes is significantly lower in the CD44+/CD24− cell (+) and ALDH1 (+) group. In subgroup analyses of the basal-like (c) and non-basal-like subtypes (d), the number of methylated genes is significantly lower in the CD44+/CD24-cell (+) group within the basal-like subtype, and there is a tendency for lower methylation in the CD44+/CD24-cell (+) group of non-basal-like subtypes.
Discussion
Although DNA methylation profiles in breast cancer have been related with breast cancer molecular subtype, there have been no studies of the association of DNA methylation with stem cell phenotype of breast cancer. In the present study, we showed that breast cancers have different methylation patterns according to molecular subtypes and that these differences were associated with the stem cell phenotypes of the tumors.
Specific oncogenic pathways have been suggested for the different breast cancer subtypes. Using array comparative genomic hybridization, candidate oncogenes have been identified in chromosomal regions 1q21–23, 10p14, and 12p13 in basal-like breast cancers, and in 1q21–23, 8p12–q21, 11q13, and 16p12–13 in luminal breast cancers.33 Moreover, it seems that DNA copy number alterations also underlie the different subtypes of breast cancer. It has been reported that higher numbers of gains/losses are associated with the basal-like subtype, while high-level DNA amplification is more frequent in the luminal B and HER2+ subtypes.34 However, not only genetic alterations but also epigenetic changes contribute to gene expression changes in the molecular subtypes of breast cancer. Previous studies have reported the association of DNA methylation patterns with molecular subtypes of breast cancer through hormone receptor and HER2/neu status.14, 15, 16, 17, 35 In an early study by Sunami et al,14 double-negative (ER-negative, HER2/neu-negative) breast cancers, corresponding to the basal-like subtype, were reported to have lower frequencies of RASSF1A, GSTP1, and APC methylation, and this was confirmed in the present study. Suijkerbuijk et al36 examined the methylation of 11 genes involved in breast carcinogenesis (RARB, RASSF1, TWIST, CCND2, ESR1, SCGB3A1, BRCA1, BRCA2, CDKN2A, APC, CDH1) by quantitative multiplex methylation-specific PCR in BRAC1-associated and sporadic breast cancers, and showed that the median values for RASSF1, TWIST, SCGB3A1, APC, and the cumulative methylation index for the 11 genes, were significantly lower in BRAC1-associated hereditary breast cancers than in sporadic ones. Similarly, Lee et al37 reported that methylation levels and ratio of SCGB3A1 (also known as HIN1), RASSF1A, and TWIST were lower in the basal-like subtype than the luminal and HER2 subtypes. As BRCA1-associated breast cancers usually are of the basal-like subtype, their observations are in agreement with our data. In the present study, the methylation frequencies and levels for 12 genes differed between subtypes and the basal-like subtype had lower methylation frequencies and levels for nine genes (APC, DLEC1, GSTP1, HOXA1, RASSF1A, RUNX3, SCGB3A1, SFRP1, and TMEFF2) than the other subtypes.
However, studies focused on individual TSGs using locus-specific methylation analysis can provide only limited information. Recent studies using array-based methylation analysis have confirmed that the molecular subtypes of breast cancer have subtype-specific methylation profiles.18, 19, 20, 21 Holm et al19 demonstrated differences in the methylation profiles of luminal A, luminal B, and basal-like breast cancers, with luminal B and basal-like breast cancers being most and least frequently methylated, respectively. In our study, the number of CpG island loci methylated was highest in the luminal–HER2 subtype and lowest in the basal-like subtype. Our data are in accord with their observation, although we only investigated a limited number of CpG islands, and subtype was defined by immunohistochemical findings of ER, PR, HER2, CK5/6, EGFR, and Ki-67. Bediaga et al18 also identified differential methylation among the tumor subtypes in 15 of 1505 screened CpG islands and found that the basal-like subtype had the lowest methylation levels among the studied subtypes.
We found that methylation of the CpG islands was lower in tumors showing stem cell phenotype as defined by CD44+/CD24− and ALDH1 expression. Basal-like tumors are known to be enriched with cells of stem cell phenotype, as shown in this study.27, 38, 39 Thus, the lower CpG island methylation in tumors of the CD44+/CD24− or ALDH1+ phenotype may reflect the association of the basal-like subtype with stem cell phenotype. However, we showed that the number of methylated genes was still significantly lower in CD44+/CD24-cell (+) tumors than in CD44+/CD24-cell (−) tumors, even within the basal-like subtype. Bloushtain-Qimrom et al28 reported that CD44+ progenitor-like cells of normal mammary epithelium were hypomethylated compared with luminal epithelial (CD24+ and MUC1+) and myoepithelial (CD10+) cells, and that cell type-specific methylation patterns were conserved in breast cancer subtypes. Kamalakaran et al20 also demonstrated that the methylation patterns in the basal subtype overlapped with those in CD44+ cells, and that the patterns in luminal subtypes overlapped with those in CD24+ cells. Taken together, these findings suggest that the distinct methylation patterns of breast cancer subtypes may reflect the methylation patterns of their different cells of origin, suggesting that basal-like tumors arise from progenitor-like cells and luminal tumors from more differentiated luminal cells.
The lower degree of methylation in the basal-like subtype is compatible with its chromosomal instability. Genomic DNA hypomethylation has been implicated in carcinogenesis by virtue of inducing chromosomal instability, as evidenced by a study in which knocking out DNA methyltransferase (DNMT) genes resulted in genomic hypomethylation and alterations of chromosome structure and/or changes in chromosome copy number.9 A recent study revealed that impaired function of BRCA1 leads to global DNA hypomethylation, loss of genomic imprinting, and an open chromatin configuration at premalignant stages of several types of tissues in a BRCA1 mutant mouse model and that in human clinical samples reduced expression of BRCA1 correlates with decreased levels of DNMT1 and reduced methylation of CpG islands.40 Thus, BRCA1 dysfunction in BRACA1-associated hereditary breast cancers and sporadic basal-like breast cancers may be associated with lower methylation of CpG islands.41, 42
We found significantly higher methylation frequencies or levels in the promoter CpG islands of nine genes (APC, DLEC1, GSTP1, HOXA1, RASSF1A, RUNX3, SCGB3A1, SFRP1, and TMEFF2) in the ER-positive group compared with the ER-negative group. An association of APC, RASSF1A, and SCGB3A1 with ER-positivity has been reported in previous studies14, 15 and our data confirmed this association. The nine genes also showed different methylation patterns in the basal-like subtype compared with other subtypes. Recently, Van der Auwera et al43 classified breast cancers into two groups with different methylation signatures (high methylation and low methylation) by unsupervised hierarchical cluster analysis of the methylation values of 500 CpG loci using 19 inflammatory breast cancers and 43 non-inflammatory breast cancer samples. DLEC1, GSTP1, RASSF1A, RUNX3, and SFRP1 were included in the genes differentially methylated in the low methylation and high methylation groups. However, they found no difference between the two groups with regard to ER, PR, and HER2 expression. This discrepancy may be due to differences in sample size, characteristics of the samples studied, and/or type of methylation analysis.
As epigenetic changes may be reversible, new therapeutic strategies focusing on epigenetic alterations have been suggested in solid tumors as well as hematolymphoid malignancies.44, 45, 46 DNMT and histone deacetylases (HDAC) have become the primary targets for the epigenetic regulation of cancer.45, 46 If DNMT and HDAC inhibitors act by reactivating TSGs, DNA methylation status may be used as a biomarker to predict response to treatment with these drugs.45 As basal-like subtype of breast cancer or breast cancer with stem cell phenotype was found to be less methylated in the genes involved in breast cancer progression, the agents that modulate epigenetic changes may be less effective in those cases. Conversely, as breast cancers with luminal–HER2 subtype or more differentiated cell phenotype show high level of promoter CpG islands methylation, they may be good targets for epigenetic therapy. Thus, distinct patterns of promoter CpG island methylation in breast cancer according to subtype or stem cell phenotype may have clinical implications in epigenetic therapy in the future.
In summary, we studied the CpG island methylation of 15 tumor-related genes with regard to breast cancer subtype, and compared the methylation patterns of the subtypes with their stem cell phenotypes. CpG island methylation was significantly lower in the basal-like subtype than in the other subtypes and the methylation of promoter CpG islands was inversely related to stem cell phenotypes of the tumor as revealed by CD44+/CD24− and ALDH1 expression. Our results suggest that breast cancers have different methylation patterns according to molecular subtype and that these are associated with stem cell phenotype of the tumors.
References
Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–752.
Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001;98:10869–10874.
Sorlie T, Wang Y, Xiao C, et al. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics 2006;7:127.
Labrie F . Drug insight: breast cancer prevention and tissue-targeted hormone replacement therapy. Nat Clin Pract Endocrinol Metab 2007;3:584–593.
Longo R, Torino F, Gasparini G . Targeted therapy of breast cancer. Curr Pharm Des 2007;13:497–517.
Beckmann MW, Niederacher D, Schnurch HG, et al. Multistep carcinogenesis of breast cancer and tumour heterogeneity. J Mol Med 1997;75:429–439.
Widschwendter M, Jones PA . DNA methylation and breast carcinogenesis. Oncogene 2002;21:5462–5482.
Costello JF, Fruhwald MC, Smiraglia DJ, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 2000;24:132–138.
Karpf AR, Matsui S . Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 2005;65:8635–8639.
Fackler MJ, McVeigh M, Evron E, et al. DNA methylation of RASSF1A, HIN-1, RAR-beta, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Int J Cancer 2003;107:970–975.
Tisserand P, Fouquet C, Barrois M, et al. Lack of HIN-1 methylation defines specific breast tumor subtypes including medullary carcinoma of the breast and BRCA1-linked tumors. Cancer Biol Ther 2003;2:559–563.
Garcia JM, Silva J, Pena C, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer 2004;41:117–124.
Park SY, Kwon HJ, Lee HE, et al. Promoter CpG island hypermethylation during breast cancer progression. Virchows Arch 2011;458:73–84.
Sunami E, Shinozaki M, Sim MS, et al. Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res 2008;10:R46.
Feng W, Shen L, Wen S, et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res 2007;9:R57.
Widschwendter M, Siegmund KD, Muller HM, et al. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res 2004;64:3807–3813.
Fiegl H, Millinger S, Goebel G, et al. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res 2006;66:29–33.
Bediaga NG, Acha-Sagredo A, Guerra I, et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res 2010;12:R77.
Holm K, Hegardt C, Staaf J, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res 2010;12:R36.
Kamalakaran S, Varadan V, Giercksky Russnes HE, et al. DNA methylation patterns in luminal breast cancers differ from non-luminal subtypes and can identify relapse risk independent of other clinical variables. Mol Oncol 2011;5:77–92.
Ronneberg JA, Fleischer T, Solvang HK, et al. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol Oncol 2011;5:61–76.
Polyak K . Breast cancer: origins and evolution. J Clin Invest 2007;117:3155–3163.
Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009;15:907–913.
Molyneux G, Geyer FC, Magnay FA, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010;7:403–417.
Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–3988.
Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555–567.
Park SY, Lee HE, Li H, et al. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res 2010;16:876–887.
Bloushtain-Qimron N, Yao J, Snyder EL, et al. Cell type-specific DNA methylation patterns in the human breast. Proc Natl Acad Sci USA 2008;105:14076–14081.
Ogino S, Kawasaki T, Brahmandam M, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn 2006;8:209–217.
Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 2010;28:2784–2795.
Hyun CL, Lee HE, Kim KS, et al. The effect of chromosome 17 polysomy on HER-2/neu status in breast cancer. J Clin Pathol 2008;61:317–321.
Voduc KD, Cheang MC, Tyldesley S, et al. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol 2010;28:1684–1691.
Adelaide J, Finetti P, Bekhouche I, et al. Integrated profiling of basal and luminal breast cancers. Cancer Res 2007;67:11565–11575.
Bergamaschi A, Kim YH, Wang P, et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer 2006;45:1033–1040.
Bertolo C, Guerrero D, Vicente F, et al. Differences and molecular immunohistochemical parameters in the subtypes of infiltrating ductal breast cancer. Am J Clin Pathol 2008;130:414–424.
Suijkerbuijk KP, Fackler MJ, Sukumar S, et al. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Ann Oncol 2008;19:1870–1874.
Lee JS, Fackler MJ, Lee JH, et al. Basal-like breast cancer displays distinct patterns of promoter methylation. Cancer Biol Ther 2010;9:1017–1024.
Honeth G, Bendahl PO, Ringner M, et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res 2008;10:R53.
Nalwoga H, Arnes JB, Wabinga H, et al. Expression of aldehyde dehydrogenase 1 (ALDH1) is associated with basal-like markers and features of aggressive tumours in African breast cancer. Br J Cancer 2010;102:369–375.
Shukla V, Coumoul X, Lahusen T, et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res 2010;20:1201–1215.
Turner NC, Reis-Filho JS, Russell AM, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2007;26:2126–2132.
Joosse SA, Brandwijk KI, Mulder L, et al. Genomic signature of BRCA1 deficiency in sporadic basal-like breast tumors. Genes Chromosomes Cancer 2011;50:71–81.
Van der Auwera I, Yu W, Suo L, et al. Array-based DNA methylation profiling for breast cancer subtype discrimination. PLoS One 2010;5:e12616.
Stearns V, Zhou Q, Davidson NE . Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest 2007;25:659–665.
Kristensen LS, Nielsen HM, Hansen LL . Epigenetics and cancer treatment. Eur J Pharmacol 2009;625:131–142.
Cai FF, Kohler C, Zhang B, et al. Epigenetic therapy for breast cancer. Int J Mol Sci 2011;12:4465–4487.
Acknowledgements
This study was supported by a Grant from Seoul National University Bundang Hospital, Republic of Korea (03-2010-002), and by a Grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (0720540), and by a Priority Research Centers Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2009-0093820).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Park, S., Kwon, H., Choi, Y. et al. Distinct patterns of promoter CpG island methylation of breast cancer subtypes are associated with stem cell phenotypes. Mod Pathol 25, 185–196 (2012). https://doi.org/10.1038/modpathol.2011.160
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.160
Keywords
This article is cited by
-
A systematic literature review assessing if genetic biomarkers are predictors for platinum-based chemotherapy response in ovarian cancer patients
European Journal of Clinical Pharmacology (2020)
-
MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer
Breast Cancer Research and Treatment (2014)
-
Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers
BMC Medical Genomics (2013)
-
The RUNX family in breast cancer: relationships with estrogen signaling
Oncogene (2013)
-
Molecular pathological epidemiology of epigenetics: emerging integrative science to analyze environment, host, and disease
Modern Pathology (2013)
