
Abstract
Histiocytic and interdigitating dendritic cell sarcomas are rare tumors originating from bone marrow-derived myeloid stem cells. Recent studies have shown evidence of cross-lineage transdifferentiation of B cells in follicular lymphoma to histiocytic and dendritic cell sarcomas. In this study, we report the morphologic, molecular and cytogenetic analysis of seven cases of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) associated with histiocytic and dendritic cell sarcomas. All seven patients were elderly males (median age 71 years). The B-cell neoplasms preceded the development of the histiocytic and dendritic cell sarcomas in six of seven patients, and one patient had both tumors diagnosed at the same time. The tumors included four interdigitating dendritic cell sarcomas: one Langerhans cell sarcoma, one histiocytic sarcoma and one immature neoplasm with evidence of histiocytic origin. Laser-capture microdissection and PCR analysis showed identical clonal immunoglobulin gene rearrangements in the two phenotypically distinct components in all cases. There was a preferential usage of IGHV4-39 by the V–D–J gene rearrangement. By fluorescence in situ hybridization (FISH) analysis, two cases showed deletion 17p in both components, whereas four cases had normal cytogenetic findings by FISH in the CLL/SLL cells, but acquired cytogenetic abnormalities in the corresponding histiocytic and dendritic tumors. Chromosome 17p abnormalities were the most common cytogenetic abnormality detected in the sarcomas, seen in five of six cases studied. Compared with the CLL/SLL cells, the histiocytic/dendritic cells were largely negative for PAX5, but showed strong expression of PU.1 and variable and weak expression of CEBPβ. Our study provides evidence for transdifferentiation of CLL/SLL B cells to tumors of dendritic and less often histiocytic lineage, and suggests that secondary genetic events may play a role in this phenomenon.
Similar content being viewed by others

Main
The WHO classification of hematopoietic neoplasms is based primarily on the lineage and stage of differentiation of the tumor cells corresponding to that of normal hematolymphoid differentiation. A characteristic feature of normal hematopoiesis is the unidirectional and irreversible lineage commitment during progressive maturation of blood cells.1 Similarly, hematopoietic tumors corresponding to mature blood cells usually demonstrate lineage stability.2 Lineage infidelity or promiscuity is often seen in precursor neoplasms, such as acute myeloid leukemia, as precursor hematopoietic cells are not fully committed to a specific lineage. Small B-cell lymphomas and leukemias recapitulate mature B cells in the pregerminal center, germinal center or postgerminal center stages of differentiation, and show the expected stability of lineage-associated markers. They have an indolent clinical course, but the most common forms, follicular lymphoma and chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), are generally incurable. During the course of disease, they may undergo transformation to diffuse large B-cell lymphoma, or even change the stage of differentiation with transformation to precursor B-lymphoblastic lymphoma,3, 4 but the B-cell lineage is usually unchanged. However, several recent studies have shown convincing evidence that some small B-cell lymphomas can transform into aggressive hematopoietic tumors of another lineage, which interestingly are of histiocytic/dendritic cell origin in all cases studied.5, 6, 7, 8
Histiocytic and dendritic cell sarcomas are extremely rare.2 By histogenesis, they fall into two main groups: histiocytic sarcoma, Langerhans cell histiocytosis/sarcoma and interdigitating dendritic cell sarcoma are thought to arise from bone marrow precursors and are considered ‘true’ hematopoietic tumors. In contrast, follicular dendritic cell sarcomas and the rarer fibroblastic reticulum cell tumors derive from non-hematopoietic mesenchymal cells. As might be anticipated, these mesenchymal neoplasms have not been linked to B-cell lymphomas. In the older literature, there are reports linking histiocytic and dendritic cell neoplasms with most subtypes of small B-cell lymphoma, including follicular lymphoma, extranodal marginal zone lymphoma (MALT lymphoma), splenic marginal zone lymphoma and CLL/SLL.9, 10, 11, 12, 13 Feldman et al7 provided the first evidence of a clonal relationship between a mature B-cell lymphoma and HD/C sarcoma in eight cases arising in follicular lymphoma. Subsequently, two other groups have demonstrated cytogenetic and molecular evidence of a clonal relationship between histiocytic or dendritic cell neoplasms and other low-grade B-cell neoplasms.5, 6, 14
In this study, we report a series of seven cases of histiocytic or interdigitating dendritic cell sarcoma arising from CLL/SLL. By molecular and cytogenetic studies, we demonstrate a clonal relationship between the two lesions. Chromosome 17p abnormalities were the most frequent cytogenetic abnormality detected by fluorescence in situ hybridization (FISH) in the sarcomas, and were also increased in frequency in the underlying CLL/SLL. Therefore, chromosome 17p changes may be a potential risk factor for the transformation of CLL/SLL to histiocytic or dendritic cell sarcoma.
Materials and methods
Case Selection
Seven cases were retrieved from the consultation files of the Hematopathology Section, Laboratory of Pathology, and Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD. Clinical information and follow-up were obtained from submitted patient records or referring physicians. Relevant clinical information is summarized in Table 1. Case 5 was previously reported as a single case report,14 but was independently analyzed in the current series. The study was approved by the institutional review board of the NCI.
Histology and Immunohistochemistry
The morphologic and immunohistochemical features were routinely studied on formalin-fixed and paraffin-embedded tissue sections. Immunohistochemical stains were performed using an automated immunostainer (Ventana Medical Systems, Tucson, AZ, USA) according to the company's protocols, with minor modifications. Antigen retrieval was performed using a Tender Cooker (Nordicware, Minneapolis, MN, USA) with citrate buffer. Antibodies included CD20 (L26, 1:1000; Dako, Carpinteria, CA, USA), CD79a (MB1, 1:400, Dako), Pax5 (24; 1:25; BD Transduction Laboratories), CD3 (F7.2.38, 1:50; Dako), CD5 (4C7, 1:100, Novocastra), CD23 (1B12, 1:40, Novocastra), CD68 (KP-1, 1:400, Dako), CD163 (10D6, 1:100, Novocastra), CD1a (O10, 1:40, Immunotech), CD4 (1F6, 1:40, Novocastra), CD21 (1F8, 1:30, Dako), CD43 (L60, 1:100, Becton-Dickinson), S100 (15E2E2, 1:8000, Biogenex), Langerin (1:200, Novocastra), PU.1 (1:50, BD Transduction), p53 (DO-7, 1:1000, Dako) and lysozyme (EC3.2.1.17, 1:2000, Dako). Positive and negative controls were performed with all cases and showed appropriate stains.
Laser-Capture Microdissection (LCM)
To compare the clonal relationship between the CLL/SLL cells and the cells of histiocytic or dendritic cell lineage, the respective neoplastic populations were isolated from the tissue sections using immuno-guided LCM technique.15, 16 Briefly, unstained sections mounted on charged glass slides were immunostained with CD79a, as described above. After staining, the slides were strongly dehydrated and no coverslip was applied. LCM was performed under microscopic visualization of the immunolabeled cells using a PixCell IIe microscope (Arcturus Engineering, Mountain View, CA, USA). Laser parameters used were: 7.5 μm laser spot size, 50 mW laser power and 950 μs laser pulse duration. Approximately between 2000 and 5000 laser pulses were completed for each sample. The cells were captured using Capsure Macro LCM Caps (Molecular Devices, Silicon Valley, CA, USA; catalog no. LCM0211). The pure populations of CLL/SLL cells were isolated by targeting only CD79a-positive small lymphoid cells, whereas pure populations of histiocytic/dendritic neoplastic cells were captured by targeting only CD79a-negative neoplastic cells. After completing the LCM, each cap with the captured cells was incubated overnight with proteinase K buffer for DNA extraction using a QIAamp DNA formalin-fixed paraffin-embedded tissue kit (QIAGEN, Valencia, CA, USA) according to the manufacturer's instructions.
PCR Studies of IGH and IGK Gene Rearrangements and Sequencing
Genomic DNA was extracted from the microdissected samples using QIAamp DNA formalin-fixed paraffin-embedded tissue kit (QIAGEN) according to the manufacturer's instructions. Given the markedly fragmented nature of genomic DNA in paraffin-embedded tissues subjected to immunohistochemical staining and LCM, FRIII IGH@ PCR was utilized to maximize the opportunity for successful amplification. For IGH@ clonality assays in cases 1, 2, 4, 5, 6 and 7, PCR was performed on each sample using framework 3 consensus primers (VHα FRIII and JHα FRIII as reported by Segal et al.17). The PCR reaction mixture contained 5 μl of DNA template purified from microdissected cells in 1 × buffer II (Applied Biosystems, Foster City, CA, USA) with 2.0 mM magnesium chloride, 0.25 mM each dNTP, 0.5 μM of each primer and 1.25 units of AmpliTaq Gold DNA polymerase (Applied Biosystems). FRII and FRIII IGH@ PCR reactions showed no amplification products in case 3. Therefore, the IGK@ clonality assay was utilized in this instance. PCR reaction using IGK Gene Clonality Assay for ABI Fluorescence Detection Kit (InVivoScribe Technologies) was performed according to the manufacturer's instructions. Briefly, 5 μl of DNA template purified from microdissected cells was mixed in 1 × IGK tube A with 1.25 units of AmpliTaq Gold DNA polymerase (Applied Biosystems). The IGK primers were originally designed by BIOMED-2 concerted action BMH4-CT98-3936.18 The DNA was amplified in a thermocycler (GeneAmp PCR System 9700, Applied Biosystems) for 7 min at 95 °C followed by 35 (for FRIII) or 40 cycles (for IGK) of amplification (95 °C for 45 s; 60 °C for 45 s; and 72 °C for 90 s) and a final cycle of extension at 72 °C for 10 min. After amplification, the PCR products were analyzed using ABI Genetic Analyzer 3130 (Applied Biosystems).
For sequence analysis, FRIII PCR reactions were performed using sequencing primers, 5′-ACACGGCTGTGTATTACTGT-3′ and 5′-ACCTGAGGAGACGGTGACC-3′ (JHα primer) in cases 1, 2, 5, 6 and 7. The PCR reaction conditions were exactly the same as described above. After PCR amplification, the PCR products were purified using the MinElute PCR purification Kit (QIAGEN) according to the manufacturer's instructions. The sequencing reaction mixture included 4 μl BigDye Terminator v3.1, 2 μl 5 × sequencing buffer (Applied Biosystems), 1 μl sequencing primer, 10 μl H2O and 3 μl of the purified PCR product. Thermal cycling condition was as follows: denaturing at 96 °C for 1 min, followed by 25 cycles at 96 °C for 10 s, 50 °C for 5 s and 60 °C for 4 min. Sequencing was done on an ABI 3100 sequencer (Applied Biosystems). The DNA sequence similarity and alignments were analyzed by CLUSTAL W 2.0.12.19 Immunoglobulin heavy chain V–D–J gene rearrangement analysis was performed using Igblast (http://www.ncbi.nlm.nih.gov/igblast/).
Fluorescence In Situ Hybridization
Formalin-fixed paraffin-embedded 5- μm sections were prepared and mounted onto positively charged glass slides. Slides were placed in a 90 °C oven for 15 min. Slides were then deparaffinized with xylene (2 times, 15 min each) at room temperature (RT), dehydrated in 100% ethanol for 5 min at RT and placed in 80 °C 10 mM citric acid (pH 6.0) for 45 min. Following this, the slides were immersed in 2 × standard saline citrate (SSC) for 5 min at 37 °C followed by digestion in 0.2% pepsin working solution (1.2 g pepsin/600 ml 0.9% NaCl, pH 2.5) at 37 °C for 48 min. Immediately after digestion, the slides were dehydrated using an ethanol series (70, 85 and 100%) for 2 min each at RT. Working solution of each probe was made by mixing 1 μl of concentrated probe with 9 μl of LSI/WCP® hybridization buffer (Abbott Molecular, Des Plaines, Ill.). Probes included centromere 6 (D6Z1) (Abbott) and c-MYB (6q23) (Mayo homebrew), centromere 11 (D11Z1) and ATM (11q22.3) (both Abbott), centromere 12 (D12Z3) (Abbott) and MDM2 (12q15) (Mayo homebrew), D13S319 (13q14.3) and LAMP1 (13q34) (both Abbott), and p53 (17p13) and centromere 17 (D17Z1) (both Abbott). The working solution was mixed together in 1:1 green to red ratio and the combined working solution was applied to the target areas, coverslipped, sealed with rubber cement, co-denatured with a HYBrite™ at 80 °C for 5 min, and hybridized overnight in a 37 °C humidified oven. Following hybridization, slides were soaked in RT 2 × SSC/0.1% NP-40 to remove coverslips, placed in 2 × SSC/0.1% NP-40 at 74 °C for 2 min and then placed into RT 2 × SSC/0.1% NP-40 for 2 min. The slides were stained with 4′-6,-diamidino-2-phenylindole (DAPI; Vysis) and coverslipped. The formalin-fixed paraffin-embedded samples were analyzed using standard fluorescence microscopy methods.20
Results
Clinical Findings
The clinical features of the seven patients are summarized in Table 1. All seven patients were male with an age range from 55 to 85 years (median 71 years). In all cases affected lymphoid tissues were involved by both CLL/SLL and a histiocytic or dendritic cell sarcoma, in different proportions, and affecting different regions. Of the seven patients, six presented originally with CLL/SLL with a subsequent diagnosis of histiocytic/dendritic sarcoma with intervals ranging from 10 months to 11 years. Patient 7 did not have a previous diagnosis of CLL/SLL, and both tumors were diagnosed concurrently. The histiocytic/dendritic sarcomas were nodal in five patients, but in two patients involved spleen (case 4) and bone marrow (case 6). Four patients (cases 3–6) received various chemotherapy regimens for CLL/SLL before the development of the secondary tumor. Regimens included fludarabine, cyclophosphamide and rituximab (case 3), cyclophosphamide and vincristine (case 4), fludarabine and rituximab (case 5) and cyclophosphamide, fludarabine, dexamethasone and rituximab (case 6). Treatment for the histiocytic/dendritic sarcomas and survival data were not available in most patients. Patient 3 received CHOP therapy for interdigitating dendritic cell sarcoma but died 4.5 months after the diagnosis.
Morphologic and Immunohistochemical Findings
In all seven cases, areas with features of CLL/SLL and histiocytic/dendritic sarcoma were present in various proportions in the same tissue. However, large confluent areas comprising CLL/SLL and histiocytic/dendritic sarcoma, respectively, were present in every case, allowing LCM of pure CLL/SLL or histiocytic/dendritic cells. The CLL/SLLs showed a diffuse growth pattern and were composed of small lymphoid cells with round nuclei and coarse chromatin. Scattered prolymphocytes and paraimmunoblasts were admixed. The CLL/SLL cells were positive for CD79a, CD5 and CD23. CD20 was negative by immunohistochemistry in two cases, which is not uncommon in CLL/SLL because of the very low level of expression of CD20.
The histiocytic/dendritic sarcomas were classified according to the criteria in 2008 WHO classification (Table 2). The tumor cells in all cases were cytologically malignant, and showed irregular nuclear contours, open chromatin, variable prominent nuclei and abundant eosinophilic cytoplasm. Except in case 7, apoptotic bodies and mitotic figures were easily seen.
Four cases (cases 1, 3, 5 and 6) were classified as interdigitating dendritic cell sarcoma. They were all strongly S100+, focally CD68+, but negative for CD163 and CD1a. They were composed of sheets of large mononuclear and multinucleated cells with abundant eosinophilic cytoplasm (Figure 1). Nuclei were highly irregular and lobulated, with distinct eosinophilic nucleoli. Mitotic figures were abundant and foci of necrosis were seen.

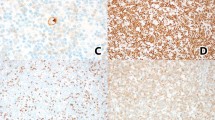
CLL/SLL and synchronous interdigitating dendritic cell sarcoma, case 3. (a) Cervical lymph node showed islands and cords of the dendritic cell neoplasm sharply segregated from areas of CLL/SLL. Magnification × 200. (b) The dendritic cells had abundant eosinophilic cytoplasm, lobulated and indented nuclei, vesicular chromatin and variably prominent nucleoli. Some cells showed highly polylobated nuclei. The CLL/SLL cells were small with round nuclei, coarse chromatin and scant cytoplasm. Magnification × 400. (c) The dendritic cells were strongly positive for S100, and partially positive for CD68. (d) They showed strong staining for PU.1 and weak and partial staining for CEBPβ, but were negative for PAX5. The CLL/SLL cells were strongly positive for PAX5 and negative for PU.1 and CEBPβ. (e) The CLL/SLL and dendritic cells showed identical clonal peaks by PCR of the IGK@ gene (IGK tube A PCR reaction). (f) Both the CLL/SLL and dendritic cell tumor showed deletion of chromosome 17p by using the FISH probe for p53 (17p13). A control probe for chromosome 17 centromere (D17Z1) showed no loss of chromosome 17.
Case 2 was classified as Langerhans cell sarcoma, being positive for both langerin and CD1a (Figure 2). It was composed of mononuclear and multinucleated cells with abundant eosinophilic cytoplasm. The nuclei had prominent nuclear grooves, often lobulated, with distinct eosinophilic nucleoli. Mitotic figures were frequent. Multiple areas of nodal involvement were seen, composed of sheets of atypical cells, with interspersed areas involved by CLL/SLL. Eosinophils were absent, arguing against a diagnosis of Langerhans cell histiocytosis.

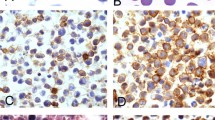
CLL/SLL and synchronous Langerhans cell sarcoma, case 2. (a) An axillary lymph node was effaced by interconnecting sheets and clusters of CLL/SLL cells admixed with large clusters of atypical Langerhans cells. Magnification × 100. The Langerhans cells were large with round to oval indented nuclei, vesicular chromatin and distinct nucleoli (inset, magnification × 400). (b) The CLL/SLL cells were positive for PAX5, CD5 and CD23. The Langerhans cells showed partial positivity for CD5, and were negative for PAX5 and CD23. (c) The Langerhans cells were positive for S100, CD1a and Langerin. (d) They showed strong staining for PU.1 and weak and partial staining for CEBPβ, but were negative for PAX5. The CLL/SLL cells were positive for PAX5 and PU.1, and negative for CEBPβ. (e) The CLL/SLL and Langerhans cells showed identical clonal peaks following IGH@ FRIII PCR (upper panel). Sequencing of IGH@ gene rearrangements (lower panel) showed complete sequence identity between that of the CLL/SLL and the Langerhans cells. The asterisks indicate identical DNA sequence. The solid lines indicate corresponding V, D and J genes. The DNA sequences between the solid lines are V–D and D–J junctional sequences. (f) The Langerhans cells showed aneusomy of chromosome 17p by using the FISH probe for p53 (17p13), and the chromosome 17 centromere (D17Z1) probe showed duplication of chromosome 17 in LCS. The CLL/SLL cells showed no abnormalities in chromosome 17.
One case (case 7) had a pure histiocytic appearance. It was composed of giant cells with voluminous pink cytoplasm, some with emperipolesis, and thus it superficially resembled sinus histiocytosis with massive lymphadenopathy (so-called Rosai–Dorfman disease; Figure 3). It was the only case that expressed both CD163 and CD68. Cytologically, the cells exhibited cellular anaplasia and atypia. Plasma cells were absent.

CLL/SLL and synchronous histiocytic sarcoma, case 7. (a) An axillary lymph node was mostly effaced by sheets of CLL/SLL cells with a focal nodule composed of atypical histiocytic cells. Magnification × 40. (b) The histiocytic sarcoma cells were very large with round nuclei, vesicular chromatin, prominent central nucleoli and voluminous pink cytoplasm. Some cells showed emperipolesis. Magnification × 100. (c) The CLL/SLL cells were positive for CD79a, CD5 and CD23. The histiocytic sarcoma cells were strongly positive for CD163. (d) They were partially positive for S100 and showed strong staining for PU.1 and CEBPβ. They were negative for PAX5. (e) The CLL/SLL and histiocytic sarcoma cells showed identical clonal peaks following IGH@ FRIII PCR (upper panel). Sequencing of the IGH@ gene rearrangements (lower panel) showed complete sequence identity between that of CLL/SLL and the histiocytic sarcoma cells. The asterisks indicate identical DNA sequence. The solid lines indicate corresponding V, D and J genes. The DNA sequences between the solid lines are V–D and D–J junctional sequences. (f) Both the CLL/SLL and IDCS showed deletion of chromosome 17p by using the FISH probe for p53 (17p13). A control probe for chromosome 17 centromere (D17Z1) showed no loss of chromosome 17. In addition, the histiocytic sarcoma showed deletion of chromosome 13q by using the FISH probe D13S319 (13q14.3). The CLL/SLL cells showed no abnormality in chromosome 13.
Case 4 was morphologically similar to cases 1, 3, 5 and 6, but at the phenotypic level was an immature neoplasm with focal expression of CD68, PU.1, CD4 and CD43, but lacking markers of more differentiated cells. It was negative for S100.
The histiocytic/dendritic tumor cells were negative for B-cell markers, such as CD20 and CD79a. Two cases (cases 5 and 6) showed patchy weak staining for PAX5 in the histiocytic/dendritic cells, readily distinguished from strong staining in the CLL/SLL cells. Similarly, cases 2 and 5 demonstrated focal weak CD5 expression in the histiocytic/dendritic cells. In all cases, the sarcomatous cells showed strong staining of PU.1 but variable and weak staining of CEBPβ. Case 7, the pure histiocytic sarcoma, had strong CEBPβ staining. As a control, the background benign histiocytes showed strong CEBPβ staining in all cases. P53 was positive in the histiocytic/dendritic tumor cells but not in the CLL/SLL cells in case 3; it was focally positive in both components of case 7.
Molecular and Cytogenetic Findings
CLL/SLL and histiocytic or dendritic tumor cells were isolated by LCM after immunohistochemical staining with CD79a and PCR analysis was performed to compare the clonal relationship between each pair of CLL/SLL and histiocytic/dendritic sarcoma. FRIII IGH@ PCR was successful in six cases, whereas case 3 amplified only with IGK@ PCR. In all cases, the CLL/SLL and histiocytic/dendritic sarcoma showed identical clonal IgH or IgK gene rearrangements by PCR (Table 3). The clonal identity was confirmed by sequencing the corresponding PCR products in five cases. Analysis of the DNA sequences revealed usage of the IGHV4-39 gene segment in four of five cases (Table 4).
FISH analysis was performed on paraffin-embedded tissue sections in six of seven cases using the CLL panel probes for detection of deletions 11q, 13q, 17p, 6p and trisomy 12 (Table 3). The CLL/SLL cells in four cases (cases 1, 2, 4 and 6) had normal cytogenetic findings, but the corresponding histiocytic/dendritic cells demonstrated a variety of cytogenetic abnormalities in 6q, 11q, 12, 13q and 17p. Two cases (3 and 7) showed deletion 17p in both the CLL/SLL and corresponding histiocytic/dendritic tumor. Chromosome 17p abnormalities, including deletion, aneusomy and duplication, were the most common abnormality observed, and were present in five of six cases (83%; cases 1, 2, 3, 6 and 7). The second most common genomic abnormality in the histiocytic/dendritic tumors was deletion 13q, which was observed in three cases (cases 1, 4 and 7).
Discussion
In recent years, histiocytic or dendritic cell neoplasms clonally related to low-grade B-cell lymphomas and leukemias have been described.7, 8, 14 This phenomenon has been attributed to lineage plasticity of the underlying B-cell neoplasm, a phenomenon first recognized in murine systems.21, 22 Before experimental data clarifying the nature of these events, there had been sporadic case reports of histiocytic or dendritic cell tumors associated with follicular lymphoma, CLL/SLL and marginal zone lymphomas.10, 12, 13 Feldman et al7 were the first to provide molecular genetic evidence of a common clonal origin for follicular lymphoma and histiocytic/dendritic sarcoma in eight cases, in which it was shown that the follicular lymphomas and histiocytic/dendritic sarcomas each shared a common IGH@ gene rearrangement and also carried the BCL2/IGH@ translocation, as shown by FISH. Thus far, a clonal relationship between CLL/SLL and a interdigitating dendritic cell sarcoma was only demonstrated in a single case report,14 and clinical or molecular risk factors for this occurrence have not been explored.
In this study, we report a series of seven patients with both CLL/SLL and a neoplasm with properties of interdigitating dendritic cells, Langerhans cells or histiocytes. All patients were elderly males. The CLL/SLL preceded the development of the histiocytic or dendritic sarcoma in six patients, and was synchronous with the secondary neoplasm in one patient. In all cases, the CLL/SLL and histiocytic/dendritic cells existed in different proportions in the same specimen. A clonal relationship between the CLL/SLL and the histiocytic/dendritic cell tumors was demonstrated by identical clonal IGH@ or IGK@ gene rearrangements in all seven patients, with the sequence identity of the corresponding V–D–J junctions confirmed in five patients. FISH analysis demonstrated chromosome 17p deletion or aneusomy in the histiocytic or dendritic cell sarcomas in five patients, and 17p deletion in the corresponding CLL/SLL in two patients.
In previous reports, the tumors associated with CLL/SLL were all interpreted as interdigitating dendritic cell sarcomas.10, 13, 14, 23, 24 Our experience is somewhat similar in that six of seven cases in our series expressed S100, and showed some evidence of dendritic cell differentiation. One of our cases expressed both CD1a and langerin and, thus, showed evidence of Langerhans cell differentiation. Transdifferentiation to dendritic cells rather than macrophages may be a preferred pathway, as dendritic cells can derive from either the common myeloid or lymphoid precursor.25 One case had a very immature phenotype, lacking convincing evidence of mature histiocytes or dendritic cells, and one case, while being focally S100+, expressed both CD163 and CD68, resembling more mature macrophages. Thus, although an interdigitating dendritic cell origin is most often seen, the secondary neoplasms can exhibit a wide spectrum of histiocytic and dendritic cell features.
A common clonal origin identified by immunoglobulin gene PCR provided strong evidence of transdifferentiation from CLL/SLL to histiocytic/dendritic cell sarcoma. Of note, sequence analysis showed preferential usage of the IGHV4-39 gene segment in the V–D–J rearrangements of the CLL/SLL and corresponding tumors. IGHV4-39 has been shown to be an independent risk factor for CLL transformation to diffuse large B-cell lymphoma.26 Thus, the use of this gene segment may be reflective of genetic instability in the B-cell clone. By FISH, all but one case of histiocytic/dendritic sarcoma had chromosomal abnormalities that were absent in the corresponding CLL/SLL, whereas no case of CLL/SLL had chromosomal abnormalities not observed in the other tumor. Thus, the FISH studies also provided indirect evidence that the development of the histiocytic/dendritic cell sarcomas was a secondary event. It would be of interest to know if the underlying CLL/SLL showed evidence of somatic hypermutation of the IGH@ genes. Unfortunately, because of the fragmented nature of the DNA obtained from the paraffin-embedded tissue, analysis of VH gene mutations could not be performed.
The events that promote the transdifferentiation of a mature B cell to a histiocyte or interdigitating dendritic cell are not well understood. However, recent data have proven the potential for continued lineage plasticity in fully differentiated cells. For example, as few as four transcription factors (Oct4, Sox2, c-Myc and Klf4) can reprogram terminally differentiated fibroblasts and hematopoietic precursor cells, including pro-B and pre-B cells, to pluripotent stem cells.27, 28, 29, 30, 31 In the hematopoietic system, deletion of the master regulator of B-cell development, PAX5, in mature B cells led to dedifferentiation into uncommitted progenitor cells and subsequent differentiation along the T-cell lineage.32, 33 Xie et al21 showed that enforced expression of transcription factors C/EBPα and C/EBPβ led to direct transdifferentiation of mature B cells to macrophages. These experiments, although all artificial with ectopic expression of higher than physiologic levels of transcription factors, suggested that lineage plasticity of mature cells can be achieved by changes of a few important transcription factors.
Two major hypotheses have been proposed for the molecular transformation of B-cell lymphoma to histiocytic or dendritic cell neoplasms. One is the direct transdifferentiation of neoplastic B cells into malignant histiocytes or dendritic cells. The other involves a two-step process of transformation with first dedifferentiation of neoplastic B cells to early progenitors and subsequent redifferentiation along the histiocytic/dendritic lineage. The latter hypothesis would require the identification of an intermediate stage that could be linked genetically to the neoplastic B cells. To date, intermediate stem cell-like forms have not been identified. However, one case in our series might represent such an intermediate step. The tumor in case 4 had a very immature phenotype, expressing CD4 and PU.1, but lacking more mature markers such as CD163, lysozyme and S100.
A third theoretical possibility would be the presence of a common neoplastic progenitor with differentiation along both B-cell and histiocytic/dendritic lineages at different times, as can be seen in chronic myelogenous leukemia. Similar to leukemic stem cells,1 such neoplastic progenitors may be mitotically inactive and resistant to chemotherapy. Dendritic cells can arise from both the common myeloid and common lymphoid progenitors.25 However, a stem cell population was not identified in our cases, making such a scenario unlikely. A common genetic marker, such as BCR/ABL, would be required to link all three stages of differentiation.
The process of differentiation is controlled by transcription factors. The B-cell fate is determined by only a few transcription factors, including E2A, EBF, PAX5 and PU.1.34, 35 Of these transcription factors, PAX5, is the single most important one for lineage commitment and maintenance of B-cell identity, and it functions throughout the life of B cells, until terminal differentiation to plasma cells.33, 36 At the final stage of B-cell differentiation, PAX5 is suppressed by Blimp1, leading to loss of B-cell identity, an event that facilitates the upregulation of genes specific for plasma cells.37, 38 As a result, mature B cells have inherent plasticity for lineage commitment during terminal differentiation to plasma cells.39 The myeloid cell fate is determined by PU.1, CEBPα and CEBPβ.40 PU.1 is involved in early multilineage differentiation and the expression levels are different in subsequent lineages.41 We tested the expression of some of the transcription factors involved in B-cell and myeloid differentiation. In all seven histiocytic or dendritic cell neoplasms, PU.1 was highly expressed, whereas PAX5 was completely negative in five cases. Interestingly, variable and patchy PAX5 expression was detected in the dendritic cell tumors in two cases. The presence of this restricted B-cell marker is consistent with the ‘transdifferentiation’ model, as incompletely transdifferentiated cells with hybrid phenotype may be identified.
The expression of CEBPβ was variable and weak in all but one case from our series, in contrast to the strong staining in histiocytic/dendritic sarcomas reported by Feldman et al.7 The weak expression was confirmed by contrasting the tumor cells with background histiocytes showing strong CEBPβ staining in the same sections. Interestingly, the one case with strong expression of CEBPβ was case 7, which showed evidence of histiocytic rather than dendritic cell features, and was more similar to the histiocytic tumors reported by Feldman et al.7
Fraser et al14 studied the expression of PU.1, CEBPα and CEBPβ by RT-PCR in a case of CLL/SLL with subsequent interdigitating dendritic cell sarcoma (also case 5 in our series). Interestingly, they showed high levels of CEBPβ by RT-PCR in both the CLL/SLL cells and the dendritic cell tumor. CEBPβ was not detected by immunohistochemistry in any case of CLL/SLL in our series, including case 5. The reasons for discrepancy in the results by RT-PCR vs immunohistochemistry are not clear, but perhaps contamination of myeloid cells in the bone marrow sample involved by CLL/SLL contributed to the RT-PCR results.
Iwama et al.42 showed that enforced PU.1 expression and impaired CEBP function promoted Langerhans cell differentiation from myeloid progenitor cells. Although the interaction of CEBP and PU.1 in interdigitating dendritic cell differentiation is not well established, the heterogeneous expression of CEBPβ coupled with the high level of PU.1 is consistent with the high incidence of tumors with Langerhans cell and interdigitating dendritic cell differentiation in our series.
The downregulation of PAX5 is crucial for loss of B-cell identity.33 Somatic mutations of PAX5 have been described in diffuse large B-cell lymphomas.43 However, in a recent study, no somatic mutations of PAX5 were identified in two cases of histiocytic sarcoma transformed from B-cell lymphoma.44 Owing to the limited quantity of microdissected DNA from paraffin-embedded tissue, we could not perform genomic PCR to assess somatic mutations of PAX5 in the current study. Inhibition of PAX5 may also be caused by chromosomal changes. However, changes in chromosome 9p13 (PAX5 gene locus) were not identified by comparative genomic hybridization by Fraser et al.14 Given that PAX5 can be suppressed by multiple transcription factors, including CEBPβ, it is likely that the initial genetic event leading to transdifferentiation of CLL/SLL involves genes other than PAX5.
In our series, the most common cytogenetic changes associated with CLL, such as deletion 13q, deletion 11q, deletion 6q and trisomy 12, were not present in the CLL/SLL cells by FISH in the six cases studied. Fraser et al14 reported trisomy 12 in both the CLL/SLL and interdigitating dendritic cell sarcoma in their case, representing case 5 in our series.14 In contrast, there were two cases of CLL/SLL with 17p13 deletion, an incidence (33%) higher than expected for de novo CLL/SLL. The 17p abnormalities were also very common in the histiocytic/dendritic sarcomas, identified in 5/6 cases studied (83%). These findings suggest that chromosome 17p abnormalities may be a potential risk factor for transformation of CLL/SLL. The 17p deletions are uncommon in de novo CLL/SLL, but when detected are associated with a more aggressive clinical course.45, 46 The two cases with 17p13 deletion in the CLL/SLL component cells (cases 3 and 7) were stained for p53. TP53 was overexpressed in the histiocytic/dendritic tumors in both cases, but in the CLL/SLL cells only in one case.
In summary, we extend the spectrum of histiocytic and dendritic cell sarcomas that may be associated with B-cell neoplasms. In CLL/SLL most secondary neoplasms show evidence of interdigitating dendritic cell differentiation, with only rare tumors composed of cells with features of mature histiocytes or macrophages. Larger series with special emphasis on the clinical course and treatment response are needed to better understand the response to different treatment strategies.
References
Chao MP, Seita J, Weissman IL . Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb Symp Quant Biol 2008;73:439–449.
Jaffe ES, Harris NL, Stein H, et al. Introduction and overview of the classification of lymphoid neoplasms. In: Swerdlow SH, Campo E, Harris NL et al. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th edn, Vol., International Agency for Research on Cancer: Lyon, France, 2008, pp 158–166.
Young KH, Xie Q, Zhou G, et al. Transformation of follicular lymphoma to precursor B-cell lymphoblastic lymphoma with c-myc gene rearrangement as a critical event. Am J Clin Pathol 2008;129:157–166.
Kroft SH, Domiati-Saad R, Finn WG, et al. Precursor B-lymphoblastic transformation of grade I follicle center lymphoma. Am J Clin Pathol 2000;113:411–418.
Wang E, Hutchinson CB, Huang Q, et al. Histiocytic sarcoma arising in indolent small B-cell lymphoma: report of two cases with molecular/genetic evidence suggestive of a ‘transdifferentiation’ during the clonal evolution. Leuk Lymphoma 2010;51:802–812.
Wang E, Papalas J, Hutchinson CB, et al. Sequential development of histiocytic sarcoma and diffuse large b-cell lymphoma in a patient with a remote history of follicular lymphoma with genotypic evidence of a clonal relationship: a divergent (bilineal) neoplastic transformation of an indolent B-cell lymphoma in a single individual. Am J Surg Pathol 2011;35:457–463.
Feldman AL, Arber DA, Pittaluga S, et al. Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008;111:5433–5439.
Zhang D, McGuirk J, Ganguly S, et al. Histiocytic/dendritic cell sarcoma arising from follicular lymphoma involving the bone: a case report and review of literature. Int J Hematol 2009;89:529–532.
Baer MR, Krantz SB, Cousar JB, et al. Malignant histiocytoses developing in patients with B-cell lymphomas. Report of two cases. Cancer 1986;57:2175–2184.
Vasef MA, Zaatari GS, Chan WC, et al. Dendritic cell tumors associated with low-grade B-cell malignancies. Report of three cases. Am J Clin Pathol 1995;104:696–701.
Wetzler M, Kurzrock R, Goodacre AM, et al. Transformation of chronic lymphocytic leukemia to lymphoma of true histiocytic type. Cancer 1995;76:609–617.
Alvaro T, Bosch R, Salvado MT, et al. True histiocytic lymphoma of the stomach associated with low-grade B-cell mucosa-associated lymphoid tissue (MALT)-type lymphoma. Am J Surg Pathol 1996;20:1406–1411.
Cossu A, Deiana A, Lissia A, et al. Synchronous interdigitating dendritic cell sarcoma and B-cell small lymphocytic lymphoma in a lymph node. Arch Pathol Lab Med 2006;130:544–547.
Fraser CR, Wang W, Gomez M, et al. Transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma to interdigitating dendritic cell sarcoma: evidence for transdifferentiation of the lymphoma clone. Am J Clin Pathol 2009;132:928–939.
Fend F, Emmert-Buck MR, Chuaqui R, et al. Immuno-LCM: laser capture microdissection of immunostained frozen sections for mRNA analysis. Am J Pathol 1999;154:61–66.
Eberle FC, Hanson JC, Killian JK, et al. Immunoguided laser assisted microdissection techniques for DNA methylation analysis of archival tissue specimens. J Mol Diagn 2010;12:394–401.
Segal GH, Jorgensen T, Masih AS, et al. Optimal primer selection for clonality assessment by polymerase chain reaction analysis: I. Low grade B-cell lymphoproliferative disorders of nonfollicular center cell type. Hum Pathol 1994;25:1269–1275.
van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003;17:2257–2317.
Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23:2947–2948.
Brockman SR, Paternoster SF, Ketterling RP, et al. New highly sensitive fluorescence in situ hybridization method to detect PML/RARA fusion in acute promyelocytic leukemia. Cancer Genet Cytogenet 2003;145:144–151.
Xie H, Ye M, Feng R, et al. Stepwise reprogramming of B cells into macrophages. Cell 2004;117:663–676.
Cobaleda C, Busslinger M . Developmental plasticity of lymphocytes. Curr Opin Immunol 2008;20:139–148.
Bonetti F, Knowles III DM, Chilosi M, et al. A distinctive cutaneous malignant neoplasm expressing the Langerhans cell phenotype. Synchronous occurrence with B-chronic lymphocytic leukemia. Cancer 1985;55:2417–2425.
Gaertner EM, Tsokos M, Derringer GA, et al. Interdigitating dendritic cell sarcoma. A report of four cases and review of the literature. Am J Clin Pathol 2001;115:589–597.
Laiosa CV, Stadtfeld M, Graf T . Determinants of lymphoid-myeloid lineage diversification. Annu Rev Immunol 2006;24:705–738.
Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol 2008;142:202–215.
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676.
Okabe M, Otsu M, Ahn DH, et al. Definitive proof for direct reprogramming of hematopoietic cells to pluripotency. Blood 2009;114:1764–1767.
Ye Z, Zhan H, Mali P, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009;114:5473–5480.
Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318:1917–1920.
Hanna J, Markoulaki S, Schorderet P, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 2008;133:250–264.
Cobaleda C, Jochum W, Busslinger M . Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 2007;449:473–477.
Carotta S, Nutt SL . Losing B cell identity. Bioessays 2008;30:203–207.
Hagman J, Lukin K . Transcription factors drive B cell development. Curr Opin Immunol 2006;18:127–134.
Ramirez J, Lukin K, Hagman J . From hematopoietic progenitors to B cells: mechanisms of lineage restriction and commitment. Curr Opin Immunol 2010;22:177–184.
Urbanek P, Wang ZQ, Fetka I, et al. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell 1994;79:901–912.
Kallies A, Nutt SL . Terminal differentiation of lymphocytes depends on Blimp-1. Curr Opin Immunol 2007;19:156–162.
Martins G, Calame K . Regulation and functions of Blimp-1 in T and B lymphocytes. Annu Rev Immunol 2008;26:133–169.
Vicente-Duenas C, Gutierrez de Diego J, Rodriguez FD, et al. The role of cellular plasticity in cancer development. Curr Med Chem 2009;16:3676–3685.
Geissmann F, Manz MG, Jung S, et al. Development of monocytes, macrophages, and dendritic cells. Science 2010;327:656–661.
Dakic A, Wu L, Nutt SL . Is PU.1 a dosage-sensitive regulator of haemopoietic lineage commitment and leukaemogenesis? Trends Immunol 2007;28:108–114.
Iwama A, Osawa M, Hirasawa R, et al. Reciprocal roles for CCAAT/enhancer binding protein (C/EBP) and PU.1 transcription factors in Langerhans cell commitment. J Exp Med 2002;195:547–558.
Pasqualucci L, Neumeister P, Goossens T, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001;412:341–346.
Bassarova A, Troen G, Fossa A, et al. Transformation of B cell lymphoma to histiocytic sarcoma: somatic mutations of PAX-5 gene with loss of expression cannot explain transdifferentiation. J Hematop 2009;2:135–141.
Zenz T, Krober A, Scherer K, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood 2008;112:3322–3329.
Zainuddin N, Murray F, Kanduri M, et al. TP53 Mutations are infrequent in newly diagnosed chronic lymphocytic leukemia. Leuk Res 2011;35:272–274.
Acknowledgements
This study was supported by the intramural research program of the Center for Cancer Research, National Cancer Institute. We thank Dr Anne Vandersteenhoven of Palmetto Richland Memorial Hospital (Columbia, SC), Dr Fan Zhou of Southwest Washington Medical Center (Vancouver, WA), Dr Stephen C Groo of Western Regional Medical Command and Madigan Army Medical Center (Tacoma, WA), Dr Wayne Tam of New York Presbyterian Hospital (New York, NY), Dr Yu Wang of St Luke's Cornwall Hospital (Newburgh, NY) and Dr Nathan A Dunsmore of Greater Baltimore Medical Center (Baltimore, MD) for their contribution of cases in this study. We also thank Thu Anh Pham and Trinh Pham in our molecular laboratory for their assistance with the molecular studies and Cynthia Harris and Hong Jin in our immunohistology lab for their help with the immunostains.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Shao, H., Xi, L., Raffeld, M. et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol 24, 1421–1432 (2011). https://doi.org/10.1038/modpathol.2011.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.102
Keywords
This article is cited by
-
Pathologic characteristics of histiocytic and dendritic cell neoplasms
Blood Research (2024)
-
Richter Transformation of Chronic Lymphocytic Leukemia—Are We Making Progress?
Current Hematologic Malignancy Reports (2023)
-
Transdifferentiation of mantle cell lymphoma into sarcoma with limited neuromuscular differentiation after conventional chemotherapy
Virchows Archiv (2022)
-
A Fibroblastic Reticular Cell Tumour Arising in the Oral Cavity: A Case Report and Review of the Literature
Head and Neck Pathology (2022)
-
The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms
Leukemia (2022)
