
Abstract
Approximately 2 billion people are infected with Mycobacterium tuberculosis (Mtb), resulting in 1.4 million deaths every year. Among Mtb-infected individuals, clinical isolates belonging to the W-Beijing lineage are increasingly prevalent, associated with drug resistance, and cause severe disease immunopathology in animal models. Therefore, it is exceedingly important to identify the immune mechanisms that mediate protection against rapidly emerging Mtb strains, such as W-Beijing lineage. IL-22 is a member of the IL-10 family of cytokines with both protective and pathological functions at mucosal surfaces. Thus far, collective data show that IL-22 deficient mice are not more susceptible to aerosolized infection with less virulent Mtb strains. Thus, in this study we addressed the functional role for the IL-22 pathway in immunity to emerging Mtb isolates, using W-Beijing lineage member, Mtb HN878 as a prototype. We show that Mtb HN878 stimulates IL-22 production in TLR2 dependent manner and IL-22 mediates protective immunity during chronic stages of Mtb HN878 infection in mice. Interestingly, IL-22-dependent pathways in both epithelial cells and macrophages mediate protective mechanisms for Mtb HN878 control. Thus, our results project a new protective role for IL-22 in emerging Mtb infections.
Similar content being viewed by others


Introduction
Approximately 2 billion people are infected with Mycobacterium tuberculosis (Mtb), resulting in 1.4 million deaths every year.1 Most infected people develop latent TB and remain asymptomatic, although they have a 10% lifetime risk of progressing into active pulmonary TB (ATB). Among Mtb-infected individuals, clinical Mtb strains belonging to the W-Beijing lineage are increasingly prevalent,2, 3 are over-represented among drug resistant isolates,4, 5 and associated with human immunodeficiency virus (HIV) infection in humans.6 In animal models, infection with Mtb HN878, a prototype of the W-Beijing lineage, is considered to be “hyper-virulent” due to increased mortality and severe disease immunopathology.7, 8 Therefore, it is important to identify the immune mechanisms that mediate protection against rapidly emerging Mtb strains, such as those belonging to the W-Beijing Mtb lineage.
The immune responses that mediate protective immunity against Mtb infections are thought to be through the production of proinflammatory cytokines such as Interferon gamma (IFNγ) and Tumor necrosis factor alpha (TNFα), cytokines that activate macrophages to mediate Mtb control.9 This is consistent with the finding that Mtb HN878 infection induces a Type I interferon response10 and T regulatory cells,8 both of which limit protective T helper type 1 (Th1) responses.8 Interleukin-17 (IL-17) is another pro-inflammatory cytokine, with a well described role in inducing cytokines and chemokines to mediate recruitment of immune cells for control of pulmonary extracellular pathogens.11 Interestingly, while IL-17 is not required for protective immunity against less virulent Mtb strains such as H37Rv,12, 13 we recently demonstrated that IL-17 is required for protective immunity against infection with Mtb HN878.14 These results bring into question whether the immune parameters required for protective immunity against emerging Mtb strains are different from immune parameters required for protection against less virulent Mtb strains.
IL-22 is a member of the IL-10 family of cytokines, and is primarily produced by T cells, γδ T cells and natural killer (NK) cells.15 IL-22 acts through its heterodimeric receptor complex composed of IL-22R1 and IL-10R2 subunits and activates signal transducer and activator of transcription 3 (STAT3) pathway.15 Although IL-22 receptor (IL-22R) is primarily expressed on epithelial cells at mucosal sites,16, 17 emerging data suggests that myeloid cells can also respond to IL-22.18, 19 IL-22 has dual roles and play protective as well as pathological functions at mucosal sites.15 For example, IL-22 overexpression can play a pathological role in several inflammatory conditions and lead to hyper-proliferation, exacerbated production of inflammatory mediators, and recruitment of pathologic effector cells.15 In contrast, IL-22 has beneficial roles in resolution of injury by promoting epithelial repair and accelerating cellular turnover,20, 21, 22 and preventing apoptosis by induction of pro-apoptotic factors such as BclIII.20 IL-22 also induces antimicrobial proteins such as β-defensins, S100A8/A9 proteins, lipocalin (Lcn) and Regenerating islet-3-gamma (Reg3γ) to mediate protection against extracellular pathogens.16, 17 Despite the extensive body of literature on the role of IL-22 at mucosal sites,15 it is unclear and controversial whether IL-22 has a protective or pathological role in immunity against Mtb. Some studies demonstrate increased IL-22 expression at disease sites in ATB patients23, 24 and in TB patients undergoing Immune Reconstitution Inflammatory Syndrome,25 suggesting a pathological role for IL-22 in TB. In contrast, other studies show increased IL-22 in serum of latent TB patients,26 and anti-tuberculosis treatment enhances IL-22 antigen-specific T cell responses in pulmonary active TB patients.27 More recently, a single-nucleotide polymorphism in the promoter region of the IL-22 gene (rs2227473) which drives increased IL-22 production and is associated with reduced susceptibility to TB,28 suggesting a protective role for IL-22 in TB. Interestingly, IL-22 treatment of Mtb infected human macrophages in vitro enhanced Mtb killing18, 29, 30 and correlated with induction of S100A8/9 proteins, Rab proteins and promoted phagolysosomal activation.18 Thus, the role for IL-22 in human TB is controversial, with some human studies suggesting a pathological role, while other studies project a protective role for IL-22 during TB. Mouse model studies published thus far have not been particularly informative, as IL-22 deficient mice are not more susceptible to aerosolized infection with less virulent Mtb strains such as H37Rv and Erdman.31, 32 Thus, in this study we addressed the functional role for IL-22 pathway in immunity to emerging Mtb strains, using Mtb HN878 as an example. Our data show that W-Beijing isolate Mtb HN878 engages Toll-like receptor 2 (TLR2) to induce increased IL-22 production and mediates protective immunity during chronic stages of Mtb HN878 infection. Interestingly, activation of IL-22-dependent pathways in both epithelial cells and macrophages together mediate protective mechanisms against Mtb HN878 infection. IL-22 functions to induce antimicrobial proteins in epithelial cells, while also inducing C–C Motif-ligand 2 (CCL2), to mediate macrophage recruitment to the infected lung during chronic disease. In addition, IL-22 can also directly activate macrophages by inducing TNFα production and mediating Mtb control. Together, these results project a new protective role for IL-22 in immunity to emerging Mtb infections, especially during chronic infection.
Results
Mtb HN878 infection drives IL-22 production through a TLR2 dependent IL-1β pathway
IL-22 deficient mice or IL-22 neutralization does not impact TB disease pathogenesis in mice infected with the less virulent Mtb strain, Mtb H37Rv.13 However, we recently found that immune requirements for protection against Mtb HN878, are not the same as following infection with the less virulent, Mtb H37Rv.14 Thus, we first tested whether IL-22 production by lung cells following Mtb infection is dependent on the infecting Mtb strain. As recently shown,14 infection with Mtb HN878 resulted in increased IL-1β production in cultured murine lung cells, when compared to infection with Mtb H37Rv (Figure 1a). We found that the induction of IL-1β is TLR2 dependent,14 as infection of Tlr2−/− in vitro cultured lung cells with either Mtb H37Rv or Mtb HN878 did not result in induction of IL-1β production (Figure 1a). Coinciding with increased IL-1β production, we found that lung cells infected with Mtb HN878 induced substantially increased levels of IL-22 protein, when compared to infection with Mtb H37Rv (Figure 1b). In addition, Mtb infection-induced IL-22 production was dependent on IL-1R signaling and TLR2 pathway (Figure 1b). Thus, our data demonstrate that hypervirulent Mtb HN878 infection induces increased IL-1β production through a TLR2 mechanism, thus driving increased IL-22 production.

Infection with Mtb HN878 triggers increased IL-17 production through a TLR2-dependent pathway. Lung cells isolated from B6, Il1r−/− and Tlr2−/− mice were infected with either Mtb H37Rv (Rv) or Mtb HN878 (HN) at multiplicity of infection (MOI) of 0.1 for 7 days, and the protein levels of (a) IL-1β and (b) IL-22 were assessed in culture supernatants by ELISA and luminex assay, respectively. Lung cells isolated from B6 mice were treated with cell wall extracted from Mtb H37Rv (Rv) or Mtb HN878 (HN), (10 μg ml−1 each). Protein levels of (c) IL-1β and (d) IL-22 were assessed by ELISA and luminex assay, respectively. PBMCs were isolated from ATB patients and were stimulated with Mtb H37Rv (Rv) or Mtb HN878 (HN) cell wall extracts (20 μg ml−1 each) and the protein levels of (e) IL-22 were measured by luminex assays. Error bars represent means±s.e.m. n=3–5 for (a–d). n=17 for human ATB patients for (e). *P⩽0.05, **P⩽0.01, ***P⩽0.001 by (a,b) one-way ANOVA, (c and d) Student’s t test (e) two-way analysis of variance. ND, not detected; IL-17, interleukin-17; TLR, Toll-like receptor; UI, uninfected; UN, untreated.
Mtb strains are characterized by differential expression of virulence factors, in particular, the complex lipid components in bacterial cell wall, which play a crucial role in host-pathogen interaction contributing to inflammatory responses.7 Mtb HN878 but not Mtb H37Rv, has an intact pks1–15 gene, which is required for the synthesis of the phenolic glycolipid (PGL), a complex branched lipid involved in host immunogenicity modulation and bacterial virulence.7 In agreement with the unique composition of mycobacterial cell wall, we found that treatment of murine lung cells with Mtb HN878 cell wall extracts induced higher production of IL-1β and IL-22, when compared to lung cells treated with cell wall extracts purified from Mtb H37Rv (Figure 1c and d). Importantly, we found that PBMCs isolated from ATB patients also responded to exposure with Mtb HN878 cell wall extracts by producing significantly increased levels of IL-22 production (Figure 1e), when compared to PBMCs stimulated with cell walls prepared from Mtb H37Rv. Thus, our data show that increased induction of IL-22 in murine cells, and human PBMCs from ATB patients is dependent on exposure to Mtb HN878 cell-wall-associated factors.
IL-22 plays a protective role during chronic stages of Mtb infection
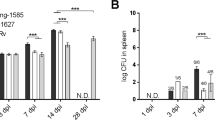
As Mtb HN878 infection in vitro induced much higher levels of IL-22 production, we then assessed if IL-22 was required for protection against Mtb HN878 infection in an aerosol mouse model of infection. While Il22−/− mice were not more susceptible during acute phases of infection, Il22−/− mice exhibited higher bacterial burden in the lungs during chronic stages whether mice were infected with either low (100 CFU) or medium (500 CFU) doses of Mtb HN878 (Figure 2a,b). The increased Mtb lung colony-forming units (CFU) during chronic Mtb HN878 infection in Il22−/− mice, also coincided with increased Mtb dissemination to the spleen in both low and medium doses of infection (Figure 2a,b). These data together suggest that the absence of IL-22 results in increased susceptibility to Mtb infection during the chronic stages of infection, but IL-22 is dispensable during the acute stages of Mtb HN878 infection.

IL-22 is required for protection during chronic stages of Mtb HN878 infection. Il22−/− and B6 mice were aerosol infected with (a) ∼100 CFU, or (b) ∼500 CFU, of Mtb HN878. Bacterial burden in the lungs and spleens was assessed by plating after (a,b) 30 and 100 days post infection (p.i). Error bars represent means±s.d. n=5–14 mice for each time point. *P⩽0.05, **P⩽0.01, ***P⩽0.001 by (a,b) Student’s t test. CFU, colony-forming units; IL-22, interleukin 22.
IL-22 signals through the receptor IL-22R, which is composed of two distinct subunits, IL-22R1 and IL-10R2.15 IL-22R is thought to be mainly expressed on non-hematopoietic cells, in particular, epithelial cells.16 However, more recent studies project macrophages as IL-22 responders.18, 29 To address the major cell type responsive to IL-22 during acute and chronic Mtb HN878 infection, we analyzed the expression of IL-22R expression in the lungs of Mtb HN878-infected mice. IL-22R expression was predominantly expressed on epithelial cells during acute stages of Mtb HN878 infection (Figure 3a). In contrast, during chronic stages of Mtb HN878 infection, IL-22R was expressed on both epithelial cells expressing E-cadherin (Figure 3b, bronchus), and recruited immune cells within TB granulomas (Figure 3a, granuloma), specifically F4/80 expressing macrophages accumulating within TB granulomas (Figure 3b, granuloma). Importantly, we found that within human TB granulomas from active TB patients, IL-22R expressing cells were predominantly CD68-expressing macrophages (Figure 3c), while IL-22R+ E-cadherin expressing epithelial cells were fewer and preferentially localized to the pulmonary epithelium (Figure 3d). These data together suggest that while IL-22 responsive cells during acute Mtb infection are primarily epithelial cells, both epithelial and accumulating macrophages within TB granulomas express IL-22R during chronic disease in mouse and human TB.

Epithelial cells and macrophages express IL-22R during Mtb infection. B6 mice were aerosol infected with ∼500 CFU Mtb HN878 and lungs were collected following acute (day 20 and 50 p.i.) or chronic (day 80 and 100 p.i.) stages of infection and formalin-fixed and paraffin-embedded (FFPE). Serial sections from FFPE sections were processed for immunofluorescence using antibodies specific for (a) CD3 (red) and IL-22R (green), (b) E-cadherin (red) or F4/80 (red) and IL-22R (green). (c) FFPE lung sections from active TB patients were processed for immunofluorescence using antibodies specific for CD68 (green) and IL-22R (red) and (d) E-cadherin (red) and IL-22R (green). All sections were counterstained with DAPI (blue). White arrows indicate cells that co-localize expression of IL-22R with CD68 or E-cadherin. n=4–6. Original magnification for a and b, × 200; c and d, left 4 × 4 × 200 mosaic; c and d, right × 200. One representative image shown. IL-22, interleukin 22.
To further address if accumulation of IL-22R-expressing macrophages is associated with Mtb control or disease progression, we assessed IL-22R expression during TB latency and TB disease in non-human primates (NHPs) infected with CDC1551, a clinical Mtb strain isolated from a TB outbreak in the United States.33 In the NHP model of TB, similar to human infection, Mtb infection can result in immune control and establishment of latent TB, or progression to TB disease. Following infection with Mtb CDC1551, we found that IL-22R-expressing macrophages were predominantly located within the non-necrotic granulomas of latently infected NHPs (Figure 4a). In contrast, IL-22R-expressing macrophage accumulation was reduced, and mainly localized on the rim of necrotic granulomas of NHPs with ATB disease (Figure 4b). We also found higher messenger RNA (mRNA) expression of IL-22R1 in TB granulomas from latently infected NHPs, when compared to IL-22R1 mRNA expression within TB granulomas from NHPs exhibiting ATB disease (Figure 4c). Interestingly, no significant induction of IL-10R2 mRNA was detected in either ATB or latently infected TB granulomas from NHPs (Figure 4c). Together, our data suggest that IL-22 responsive macrophages accumulate during infection with clinical Mtb strains and may participate in immune protective control during TB.

Preferential accumulation of IL-22+ macrophages within granulomas of latent tuberculosis (TB). Non-human primates (NHPs) were aerosol infected with Mtb CDC1551 and lungs were collected from NHPs with (a) latent and (b) acute TB. Formalin-fixed and paraffin-embedded (FFPE) lung sections were prepared and stained for (left) H&E or immunofluorescence with antibodies specific for (middle) E-cadherin (red) and IL-22R (green), or (right) macrophage (green) and IL-22R (red). All sections were counterstained with DAPI (blue). White arrows indicate co-localization of IL-22R with macrophage or epithelial markers. Original magnification for (left) H& E panel is × 100, (middle and right) immunofluorescence panels × 200. (c) Induction of IL-22RA1 and IL-10RA was measured by real-time PCR and fold induction in either latently or actively infected NHP lungs over levels expressed in control lungs is shown. n=4–6. Error bars represent means±s.e.m. IL, interleukin; ND, not detected.
IL-22 is required for induction of Reg3γ and anti-mycobacterial control in epithelial cells
We found that both macrophages and epithelial cells express IL-22R during Mtb infection in animal models and human TB granulomas. Thus, we first assessed if the epithelial barrier was dysregulated in the absence of IL-22 during chronic Mtb HN878 infection. We observed that while lungs from B6 mice infected with either low or medium doses of Mtb HN878 during the chronic stages, exhibited well maintained epithelial barrier integrity, the epithelial barrier in the lungs of Il22−/− Mtb HN878-infected mice was aberrant and discontinuous (Figure 5a and b). In addition, we found that the mRNA induction of IL-22-dependent antimicrobial genes, such as S100A8 and Reg3γ, was decreased in the lungs of medium dose Il22−/− Mtb HN878-infected mice, when compared to B6 Mtb HN878-infected mice, while expression of Lcn2 mRNA was not significantly different (Figure 5c). Interestingly, these differences in epithelial barrier integrity or decreased induction of IL-22-dependent antimicrobial genes did not impact overall inflammation associated with the chronic infection in medium dose Il22−/− Mtb HN878-infected mice, when compared to inflammatory changes in lungs of B6 Mtb HN878-infected mice (Figure 5d). To further address if IL-22 has a direct anti-mycobacterial function at the epithelial barrier, we treated non-tumorigenic lung epithelial cell line, C10, with recombinant (r) IL-22 and found that this resulted in mRNA induction of antimicrobial proteins including Reg3γ, S100A8 proteins and Lcn2 (Figure 5e). Furthermore, we found that pre-treatment of C10 cells with rIL-22, resulted in improved Mtb HN878 control by epithelial cells (Figure 5f). Consistent with improved Mtb control upon rIL-22 treatment, rIL-22 pre-treatment also decreased Mtb HN878 uptake in C10 cells (Figure 5g). Additionally, we found that rReg3γtreatment of Mtb HN878-infected C10 cells also resulted in improved Mtb HN878 control, albeit not to the extent seen with rIL-22 treatment (Figure 5h). These data together suggest during the chronic stages of Mtb HN878 infection, IL-22 plays an important in epithelial barrier structure and function, likely through the induction of antimicrobials.

IL-22 regulates epithelial cell function following infection with Mtb HN878. Il22−/− and B6 mice were aerosol infected with a 100 or 500 CFU Mtb HN878 and on day 100 p.i, lungs were collected. FFPE lung sections were processed for performing (a) H&E staining and immunofluorescent staining with antibodies specific for E-cadherin (red). All sections were counterstained with DAPI (blue). Circles indicate areas shown for immunofluorescent images. (b) Quantification of E-cadherin from lung FFPE sections was determined measuring the area covered by E-cadherin fluorescence in medium size bronchi (<150 μm in diameter). (c) Induction of Reg3γ and S100A8 mRNA was measured by RT-PCR and fold induction of mRNA shown. (d) Percentage of granulomatous inflammation in the lungs was assessed using an automated tool of the Zeiss microscope. (e) C10 cells were incubated with rIL-22 (100 ng ml−1) and induction of Reg3γ, S100A8 and Lcn2 mRNA was measured by real-time PCR and fold induction over untreated cells is shown. C10 cells (6 × 105–9 × 105) were infected with Mtb HN878 (multiplicity of infection (MOI) of 1) and treated with either (f) rIL-22 (100 ng ml−1) or (g) recombinant cleaved Reg3γ (25 μg ml−1) and bacterial burden assessed at 6 days following infection. n=4–6. Error bars represent ±s.d. (h) C10 cells were pretreated with rIL-22 (100 ng/ml) for 48 h and then infected with HN878:GFP at a MOI of 1 (5 × 105 C10 cells) for an additional 4 h, and assessed for percent Mtb:GFP+ uptake. *P⩽0.05, **P⩽0.01, ***P⩽0.001 by (c,d,f–h) Student’s t test. GFP, green fluorescent protein; NS, not significant; ND, not detected; UN, untreated.
IL-22 is required for macrophage recruitment during chronic Mtb HN878 infection
Our new data show that macrophages are one of the predominant recruited cell types expressing IL-22R during chronic Mtb infection in animal models and human TB granulomas. Thus, we next addressed whether the accumulation of recruited macrophages were impacted in Il22−/− Mtb HN878-infected mice during either acute or chronic stages of Mtb infection. We found that following low dose (Supplementary Figure 1a–c online) or medium dose (Figure 6a–c) Mtb HN878 infection, the number of alveolar macrophages, monocytes and recruited macrophages in the lungs were comparable between B6 and Il22−/− mice during acute stages of Mtb HN878 infection. In contrast, following both low and medium doses of Mtb HN878 infection, we observed decreased number of recruited macrophages accumulating within Il22−/− Mtb HN878-infected lungs, when compared to wild-type infected lungs. In addition, during the chronic stages, alveolar macrophage numbers were reduced in infected Il22−/− mice following low dose Mtb HN878 infection (Supplementary Figure1a), while monocytes were reduced in medium dose Mtb HN878-infected Il22−/− mice (Figure 6b). Neutrophilic accumulation at low or medium dose Mtb HN878 infection during acute and chronic stages of infection was not statistically different between wild type and Il22−/− -infected lungs (Figure 6d and Supplementary Figure 1d). These results were further confirmed histologically, as decreased accumulation of IL-22R+-expressing F4/80 macrophages were observed within TB granulomas in chronically infected Il22−/− mice, irrespective of whether mice were infected with low or medium dose Mtb HN878 (Figure 6e,f). The number of IFNγ-producing activated CD4+ and CD8+ T cells in Il22−/− mice infected with medium doses of Mtb HN878 show increases during acute infection but no significant difference when compared to B6 during chronic stages of infection (Supplementary Figure 2). In contrast, IL-17+ activated CD8 T cells in Il22−/− mice was increased during acute and chronic stages of infection (Supplementary Figure 2). Similar results were observed during low-dose Mtb HN878 infection of Il22−/− mice (data not shown). Together, our data indicate that Il22−/− mice infected with Mtb HN878 show defects in recruitment and accumulation of IL-22R-expressing macrophages within TB granulomas during chronic Mtb infection.

IL-22 is required for macrophage and monocyte recruitment during the chronic stages of Mtb HN878 infection. Il22−/− and B6 mice were aerosol infected with ∼500 CFU of Mtb HN878 and lungs collected at different time points p.i. The total number of (a) alveolar macrophages (AMs), (b) monocytes (Mn), (c) recruited macrophages (RM), (d) neutrophils (Neu) were determined by flow cytometry. (e) FFPE lung sections were processed for lung pathology and immunofluorescence using antibodies specific for F4/80 (red) and IL-22R (green). All sections were counterstained with DAPI (blue). Circles indicate region of interest for F4/80+ and IL-22R+ within the granulomas. (f) Quantification of IL22R+ cells in lung sections from (e) were counted /200x field. Error bars represent (±s.d. n=five mice, Representative individual experiments are shown from at least two experiments. **P<0.01, ***P<0.001 by Student’s t test. CFU, colony-forming units; IL, interleukin.
To determine the cellular source of IL-22 following Mtb HN878 infection, we infected total lung cells in vitro with Mtb HN878 and found that while both CD4+ and CD8+ cells are the primary producers of IL22 (Supplementary Figure 3a), innate cells such as natural killer (NK) cells, NK T cells, and TCRγδ cells also produce IL-22 (Supplementary Figure 3b). Thus, our data suggest that both innate cells as well as adaptive cells can produce IL-22 in response to Mtb HN878 infection.
We next addressed whether IL-22 can act on epithelial cells in order to induce chemokines that mediate macrophage recruitment into granulomas. We treated C10 epithelial cells with rIL-22, and observed that along with induction of antimicrobial proteins (Figure 5e), rIL-22 treatment resulted in induction of mRNA and protein levels of several chemokines including neutrophil-attracting chemokines, such as CXCL1, CXCL5 and G-CSF in epithelial cells (Figure 7a,b). Importantly, we also found that rIL-22 treatment of C10 cells induced CCL2, a macrophage recruiting chemokine in TB34 (Figure 7a,b). Coincident with ability of rIL-22 to induce CCL2, we also found decreased CCL2 mRNA localization within lungs of Il22−/− Mtb HN878-infected mice, when compared to lungs of B6 Mtb HN878-infected during chronic stages of infection (Figure 7c). These data suggest that IL-22 can act on epithelial cells to induce both neutrophil and macrophage attracting chemokines. These findings are consistent with the reduced recruitment of macrophages during chronic infection in Mtb HN878-infected Il22−/− mice, suggesting an important role for IL-22 in myeloid cell recruitment during chronic TB.

IL-22 activates macrophages for Mtb control. Non-tumorigenic lung epithelial cell line C10 was treated with rIL-22 and (a) chemokines mRNA was determined by real-time PCR, and fold induction over untreated cells was determined or (b) protein levels were determined in culture supernatants by luminex assays. (c) Il22−/− and B6 mice were aerosol infected with ∼500 CFU of Mtb HN878 and on day 100 p.i., formalin-fixed, paraffin-embedded lung sections were analyzed by ISH to determine localization of CCL2 mRNA expression within the lung. Black arrows indicate the localization of CCL2 mRNA within granulomas. Original magnification, × 10. BMDMs derived from B6 mice were treated with rIL-22 and infected with Mtb HN878 (multiplicity of infection (MOI) of 1) and (d) intracellular Mtb burden was determined by plating and (e) the protein levels of TNFα production was determined in culture supernatants by ELISA. n=4–6. (f) B6 macrophages were pretreated with rIL-22 (100 ng ml−1) for 48 h and then infected with HN878:GFP at an MOI of 1 for an additional 4 h, percent Mtb:GFP uptake was assessed. *P⩽0.05, **P⩽0.01, ***P⩽0.001 by (d,e) one-way analysis of variance or (b,f) students t-test. Error bars represent s.d. Representative individual experiments are shown from at least two experiments. ND, not detected; UI, uninfected; UN, untreated; NS, not significant.
To further understand whether IL-22 can directly modulate macrophage function during Mtb infection,18 we then treated Mtb-infected macrophages with rIL-22 and assessed intracellular Mtb control. Similar to rIFNγ treatment of macrophages, we found that rIL-22 treatment also decreased intracellular Mtb burden in macrophages (Figure 7d). Similar to rIFNγ treatment of BMDMs, improved Mtb control in rIL-22 treated Mtb-infected macrophages coincided with increased TNFα production (Figure 7e). In contrast, rIL-22 treatment did not affect Mtb HN878 uptake by macrophages (Figure 7f). These data suggest that IL-22 directly induces TNFα to activate macrophages, enhancing intracellular killing mechanisms to control Mtb growth. Thus, our novel data show that IL-22 induces chemokine production in epithelial cells to recruit monocytes and macrophages, and stimulates IL-22-responsive macrophages to directly induce TNFα for macrophage activation and Mtb control.
Discussion
In the present study, we investigated the functional role of IL-22 in host immune responses to infection with emerging “hyper-virulent” Mtb HN878. Unlike infection with less virulent Mtb H37Rv infection,13 we report that IL-22 promotes protective immunity during chronic stages of Mtb HN878 infection. We show that IL-22 acts on lung epithelial cells to induce antimicrobial proteins as well as monocyte and macrophage recruiting chemokines like CCL2, to mediate control of Mtb HN878. In addition, we show that IL-22R is expressed on macrophages that accumulate within lung TB granulomas, and that IL-22 can directly induce TNFα production and activate macrophages to mediate mycobacterial control. Together, our results describe a functional role for IL-22 in mediating protective immunity to TB, with involvement from both epithelial cells and macrophages in mediating IL-22-dependent protective mechanisms in TB. Thus, our results demonstrate that immune requirement for emerging clinically relevant Mtb strains such as HN878, may be different from those required for control of lab-adapted Mtb strains such as H37Rv.
When compared to infection with Mtb H37Rv, our data show that Mtb HN878 infection of murine lung cells is more potent at inducing IL-1β and IL-22 production. In addition, the induction of IL-22 in lung cells in response to Mtb HN878 infection is dependent on IL-1R pathway, projecting increased IL-1β induction by Mtb HN878 as a critical step in driving increased production of both IL-1714 as shown before, and IL-22, as shown here. Mtb HN878 uniquely expresses phenolic glycolipids (PGLs), a major lipid component of the mycobacterial cell wall.7, 10, 35 Increased induction of both IL-1β and IL-22 production upon exposure to cell wall components of Mtb HN878 in murine and human cells, suggests that production of PGLs by Mtb HN878 may be a possible pathway for induction of high levels of IL-22. Future studies delineating the molecular host-pathogen interactions that mediate differential induction of IL-22 following infection with different Mtb lineages would thus be critical in fully understanding the role of IL-22 in TB.
The low induction of IL-22 in response to Mtb H37Rv infection in vitro, coincides with a redundant role for IL-22 in mediating protective immunity against less virulent Mtb strains.13, 31 Accordingly, following induction of high levels of IL-22 in response to Mtb HN878 infection in vitro, we show that IL-22 plays an important role in mediating protective immunity to Mtb HN878 infection during chronic infection in mice. Interestingly, Il22−/− mice are not more susceptible to Mtb HN878 during acute stages of Mtb HN878 infection, but show increased susceptibility during the chronic stages of infection. This is in contrast to Il17−/− mice that show increased susceptibility during acute stages of Mtb HN878 infection.14 Our published studies show that Il17−/− mice have reduced CXCL13 mRNA localization in the lung, decreased formation of lymphoid containing granulomas and decreased activation of macrophages in Il17−/− Mtb HN878 infected mice.14 Thus, it is possible that early increased IL-17 induced in the lung is sufficient to maintain protective immunity during acute stages of Mtb HN878 infection in Il22−/− mice. Interestingly, and in contrast to a protective role for IL-17 during early stages of Mtb HN878 infection,14 IL-17 production in T cells during chronic infection in Il22−/− mice does not appear to be sufficient for protective immunity. Furthermore, while we did not observe differences in overall lung inflammation in B6 and Il22−/− Mtb-HN878 infected mice, we observed both dysregulated epithelial barrier function and decreased macrophage recruitment in chronically infected Il22−/− mice. Interestingly, infection of epithelial cells with Mtb HN878 results in greater uptake and infection of epithelial cells,36 suggesting that the epithelial barrier may have an important role to play in protective immunity to emerging Mtb strains. It is interesting that treatment of epithelial cells with rIL-22 can reduce uptake and control Mtb HN878 growth in epithelial cells. Similarly, treatment with Reg3γ can also reduce Mtb HN878 growth in epithelial cells, albeit not to levels induced by rIL-22 treatment. These results suggest that IL-22 dependent induction of antimicrobial peptides including Reg3γ, Lcn2 and S100A8 proteins can likely mediate direct anti-mycobacterial control at the epithelial barrier. A role for IL-22 during chronic stages of Mtb infection coincides with decreased induction of CCL2 mRNA within TB granulomas and associated reduction in recruitment of macrophages within TB granulomas. IL-22 treatment of epithelial cells also drives induction of G-CSF and chemokines associated with neutrophil responses. However, as differences in neutrophilic accumulation during chronic stages of Mtb HN878 infection in Il22−/− and B6 mice were not dramatic, we predict that IL-17 may be playing a compensatory role in driving neutrophil responses in the Il22−/− infected mice. It is thus likely that the major protective effects of IL-22 in vivo during chronic infection are mediated through the accumulation of newly recruited macrophages, rather than through direct anti-mycobacterial mechanisms at the epithelial barrier or neutrophil mediated control. The future availability of IL-22R deficient floxed mice that specifically lack IL-22R expression either on epithelial cells or macrophages and neutrophils will allow us to define the role of epithelial cell versus myeloid cells in IL-22-dependent immunity to TB.
IL-22R is thought to be expressed primarily on epithelial cells at mucosal sites.16, 17 However, emerging evidence projects myeloid cells as novel responders to IL-22.18, 19 We found that during the acute stages of Mtb HN878 infection in mice, IL-22R was primarily expressed on epithelial cells. However, during progression into chronic stages of Mtb HN878 infection in mice, we observed localization of F4/80-expressing macrophages that also expressed IL-22R within TB granulomas. Importantly, we also observed that the predominant cells expressing IL-22R within human TB granulomas and latently infected NHPs was also CD68-expressing macrophages. As NHPs infected with Mtb CDC1551, and mice infected with Mtb HN878, both exhibited accumulation of IL-22R-expressing macrophages within TB granulomas, suggesting that the IL-22R-expressing macrophage accumulation with TB granulomas is a more general response to Mtb infection with clinical strains. These data are consistent with direct IL-22-dependent induction of TNFα in macrophages, and improved intracellular Mtb control. Previous studies have also shown that IL-22 can act on Mtb-infected human macrophages to induce S100A8/A9 proteins, enhance phagolysosomal fusion and improve Mtb killing.18, 30 Thus, IL-22 has an emerging role in activating macrophages to mediate anti-mycobacterial killing mechanisms to mediate protective immunity to TB.
Increased IL-22 expression is observed at disease sites in ATB patients,23, 24 and accumulation of IL-22-producing T cells have been documented within TB granulomas in NHPs.37 In addition, both innate and adaptive immune cells produce IL-22, as NK 1.1+ cells,29, 38 CD4+ T cells23, 26 and granulocytes26 have all been reported to produce IL-22 during TB. This is consistent with our studies which show that both innate and adaptive cells in the lung produce IL-22 upon Mtb HN878 infection. On the basis of our results, we thus speculate that increased level of IL-22 observed in serum of latent TB patients26 functions protectively and is able to limit disease progression. However, overexpression of IL-22 in pulmonary disease sites23, 24 may mediate exacerbated pathology during active TB. Considering that anti-tuberculosis treatment enhances IL-22 antigen-specific T-cell responses in ATB patients,27 it is possible that enhancing IL-22 T cell responses by vaccine strategies will benefit host immunity to control Mtb. Furthermore, as identification of a single-nucleotide polymorphism that drives increased IL-22 production in humans is associated with reduced susceptibility to TB,28 our results together project a protective role for IL-22 in TB.
In conclusion, in the current study we demonstrate a novel role for IL-22 in mediating protective immunity to TB, especially following infection with emerging Mtb strains, specifically at the chronic stages of infection. Interestingly, IL-22-dependent pathways in both epithelial cells and macrophages together mediate protective mechanisms against Mtb HN878 infection. Together, these results project a novel protective role for IL-22 in emerging Mtb infections.
Methods
Human tissue samples and patient diagnosis. Archival paraffin samples of human lung biopsies with diagnosis of TB were collected under a protocol approved by the Ethics Committee of the National Institute for Respiratory Diseases (INER), Mexico. Subjects were of similar socioeconomic status and unrelated to the third generation as determined by a questionnaire. TB patients had evident symptoms (weight loss >10 kg, cough, fever, night sweats for >1 month, or cervical or axillary lymphadenopathy) and chest radiographic findings consistent with recent pulmonary TB. TB was confirmed with a positive sputum acid-fast smear and culture. Fresh blood samples from ATB patients were obtained from patients recruited to the Tuberculosis Outpatient Clinic, INER, Mexico prior to anti-Mtb treatment and did not present co-morbidities such as diabetes, HIV, cancer and Chronic Obstructive Pulmonary Disease. Human peripheral blood mononuclear cells (PBMCs) from ATB patients were isolated by Ficoll Hypaque gradient (Lymphoprep; Axis-Shield POC AS, Oslo, Norway). PBMCs (2.5 × 106 cells per ml) were plated in 24-well plates and incubated at 37 °C in presence of cell wall extract (20 μg ml−1) from Mtb H37Rv or Mtb HN878. After a 4-day incubation period, cells were collected and culture supernatants were used to determinate the levels of IL-22 (Bio-Rad Laboratories, Hercules, CA, USA).
Mtb infection in non-human primates (NHPs). Groups of NHPs (Indian rhesus macaques) were used in this study at the Tulane National Primate Research Centre as previously described following the recommendations of the Institutional Animal Care and Use Committee. For latently infected NHPs, NHPs were infected with a low dose of Mtb CDC1551 (200–500 CFU) by the aerosol route.39 Mtb CDC1551 exposure was confirmed in NHPs by positive tuberculin skin test and PRIMAGAM (Prionics), a quantiferon-type IFNγ release assay specifically designed to confirm Mtb infection in NHPs. The NHPs with latency did not exhibit any clinical signs of TB, as evident from normal body temperatures and weights, chest X-rays and serum levels of acute phase proteins.40 For pulmonary TB in NHPs, NHPs were infected with a high dose of Mtb CDC1551 via aerosol infection (∼5,000 CFU). Mtb exposure was confirmed by positive tuberculin skin test and PRIMAGAM and the NHPs exhibited clinical signs of TB, as evident from body temperatures, weights and chest x-rays.40 Depending on disease severity, NHPs with ATB were necropsied at different time points with the average time to euthanasia being 37 days. Latently infected NHPs were necropsied at varying time points due to experimental and ethical considerations rather than disease severity (as latently infected NHPs are otherwise normal), and the average time to necropsy was 141 days.
Mice. C57BL/6 (B6), Il1r−/−, Tlr2−/− on the B6 background were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred at Washington University in St Louis animal facility. Il22−/− mice16 were obtained from Dr Wenjun Ouyang and bred in-house. Experimental mice were age- and sex-matched and used between the ages of 6–8 weeks. All mice were maintained and used in accordance with the approved Institutional Animal Care and Use Committee (IACUC) guidelines at Washington University in St Louis.
Bacterial strains and cell wall preparations. Mtb strains H37Rv (Trudeau Institute) and HN878 (BEI Resources, and a kind gift from Dr Gilla Kaplan, PHRI) were cultured in Proskauer Beck medium containing 0.05% Tween 80 to mid-log phase and frozen in 2 ml aliquots at −80 °C. Mice were aerosol infected with approximately 100 or 500 Mtb CFU as before.14 At given time points, organs were collected, homogenized and serial dilutions of tissue homogenates plated on 7H11 agar plates and CFU determined.
H37Rv (NR14828) or HN878 (NR14830) cell wall preparations were commercially obtained from BEI Resources, under National Institutes of Health (NIH) contract AI-75320. Mtb cultures were collected at late-log phase in glycerol-alanine-salts medium, washed with PBS pH 7.4, and subsequently inactivated by gamma irradiation. The bacilli were broken in a French Press pressure cell at 4 °C. The cell wall was isolated by centrifugation at 27,000 × g for one hour and washed two times in PBS. The final cell wall pellet was suspended and dialyzed in 10 mM ammonium bicarbonate, quantified by BCA protein assay for protein content.
Preparation of bone marrow-derived macrophages. BMDMs were generated from the bone marrow of B6 or gene-deficient mice as described previously.14 On day 7, adherent cells were collected and used as BMDMs which were used to perform either in vitro infection or treatment with bacterial cell wall preparations (10 μg ml−1 each, BEI Resources, obtained under National Institutes of Health (NIH) contract AI-75320).
Preparation of lung cell suspensions. Lung cell suspension was prepared from the lungs of B6 and gene deficient mice as described previously.14 Briefly, lungs were taken and perfused with cold saline containing heparin prior to sectioning in chilled DMEM using sterile razor blades. Dissected lung tissue was then incubated in DMEM containing collagenase (0.7 mg ml−1; Sigma-Aldrich) and DNase (30 μg ml−1; Sigma-Aldrich) at 37 °C, 7.5% CO2 for 30 min. Digested lung tissue was dispersed by passage through a 70 μm pore size nylon tissue strainer (Falcon; BD Biosciences, San Jose, CA). Single cell suspension was treated with Gey’s solution to lyse any residual red blood cells and washed twice before counting.
In vitro Mtb infection. BMDMs and total lung cells were prepared as described above and plated at a density of 1 × 106 cells per ml and rested at 37 °C, 7.5% CO2 overnight. Non-tumorigenic lung epithelial cell line, C10 was cultured to 100% confluence prior to infection or pretreatment with recombinant IL-22 (R&D Biosystems, Minneapolis, MN), or the cleaved form of Reg3γ.41 A representative well was taken for counting using a haemocytometer and to calculate the multiplicity of infection (MOI). Cells were subsequently infected with Mtb HN878 at a MOI of 1 for BMDMs and C10 cells, and MOI of 0.1 for lung cells. Infected cells were cultured in antibiotic-free DMEM (with 10% fetal bovine serum) at 37 °C, 7.5% CO2 for 6 days. Plates were then centrifuged, and culture supernatant stored at −80 °C for further analysis. The infected BMDMs were washed once with sterile PBS and lysed with 0.05% SDS, following which serial dilutions were plated on 7H11 plates and CFU determined.14
Following a 48 h pretreatment with rIL-22 (100 ng ml−1), BMDMs or C10 cells were assessed for mycobacterial uptake using Mtb HN878:GFP for infection at a MOI of 1 for 4 h. After removal of the supernatants by centrifugation, single cell suspensions of infected cells were generated using.25% trypsin (Gibco) treatment for 4 min followed by equal volume of DMEM (with 10% fetal bovine serum). Suspensions were analyzed for GFP+ cells against uninfected controls using the BD FACs Jazz cell sorter and post run analysis using FlowJo (Tree Star, Ashland, OR). Data are reported as %Mtb-GFP uptake.
Flow cytometry. Lung cell suspensions were prepared, stained, collected and analyzed for flow cytometry as described earlier.12 Fluorochrome-labeled antibodies specific for CD3 (500A2), CD4 (RM4–5), CB8α (53-6.7), CD44 (IM7), IFNγ (XMG1.2), IL-17 (TC11-18H10), IL-22 (1H8PWSR), CD11b (M1/70), CD11c (HL3), NK1.1 (PK136), TCRγδ (GL3) and Gr-1 (RB6-8C5), or isotype control antibodies were used in this study. Cells were collected using a Fortessa X20 Cytometer with FACS Diva software. Cell populations were gated based on their forward by side scatter characteristics and the frequency of specific cell types was calculated using FlowJo (Tree Star). Lung alveolar macrophages were gated as CD11c+ CD11b−, lung myeloid dendritic cells were gated on CD11c+ CD11b+, neutrophils as CD11b+ Gr-1hi, recruited macrophages were annotated as CD11b+ Gr-1lo, and monocytes were gated on CD11b+ Gr1int cells as before.42
Lung morphometric analysis and immunofluorescence. Mouse lung lobes were processed as described previously.12 Lung sections were stained with hematoxylin and eosin (H&E) and inflammatory features were blinded and evaluated by light microscopy. Formalin-fixed lung sections were cut, stained and performed immunofluorescence analysis was performed as described previously.14 Regions of inflammatory accumulation were delineated with the automated tool of the Zeiss Axioplan 2 microscope (Carl Zeiss, Thornwood, NY) and average size in squared microns calculated. Sections were probed with F4/80 (MCA497GA, Serotec, Hercules, CA) to detect activated macrophages within inflammatory lesions, and goat anti-mouse E-cadherin (AF748, R&D Systems, Minneapolis, MN) to detect epithelial cells. Quantification of E-cadherin from lung FFPE sections was determined measuring the area covered by E-cadherin fluorescence in medium size bronchi (<150 μm in diameter) using the automated tool of the Zeiss Axioplan 2 microscope (Carl Zeiss). Rabbit anti-mouse IL-22Rα (ab5984, Abcam, Cambridge, MA) or rat anti-mouse IL-22Rα1 (clone 496514, R&D systems) was used to visualize IL-22R expression. Non-human primate (NHP) and human sections were probed with mouse anti-human macrophages (clone MAC387, AbD Serotec) to detect activated macrophages within inflammatory lesions, anti-human cadherin (AF648, R&D systems) to stain for epithelial cells, mouse monoclonal anti-human IL22R alpha 1 (MAB2770, R&D systems) to observe IL-22R expression or mouse monoclonal anti-human CD68/Macrosialin (clone PG-M1, Thermo Fisher Scientific, Waltham, MA) for evaluating macrophage accumulation in infected lungs.
Real-time PCR. RNA was extracted and cDNA was generated as previously described.12 cDNA was then amplified using TaqMan reagents on the Applied Biosystems ViiA 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). The log10 fold induction of mRNA in experimental samples was calculated over expression levels in uninfected samples was determined using the ΔΔct calculation recommended by manufacturer. The primer and probe sequences for murine glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were previously published.14 The primer and probes for murine Reg3γ, S100A8, Lcn2, CCL2, chemokine (C-X-C motif) ligand 1 (CXCL1), granulocyte-colony stimulating factor (G-CSF) and IL-22R were purchased from Applied Biosystems.
Quantification of protein production. Mouse IL-1β ELISA set (BD Biosciences, San Jose, CA) and mouse TNFα ELISPOT pair (BD Biosciences) was used to detect IL-1β and TNFα protein levels in the supernatant samples according to the manufacturer’s protocol. IL-22, G-CSF and chemokine protein levels were measured using a mouse Luminex assay (Linco/Millipore, Bedford, MA).
In situ hybridization (ISH). Paraffin embedded tissue sections were deparaffinized and prepared for performing in situ hybridization as previously described.43 In brief, stringent in situ hybridization (50 °C with 0.1 M DTT in the hybridization mix) was performed with 35S-labeled riboprobes as previously described.43 Tissue sections were coated with NTB emulsion (Carestream/Kodak, Rochester, NY), exposed at 10 °C for 14 days, counterstained with hematoxylin (Vector Laboratories, Burlingame, CA) and mounted with Permount (Fisher, Waltham, MA). Images were visualized using Leica DMC2900 and captured using LAS V4.5 program (Diagnostic Instruments, Sterling Heights, MI).
Statistical analysis. Differences between the means of two groups were analyzed using the two tailed Student’s t test or between multiple groups using one- or two-way ANOVA in GraphPad Prism 5 (La Jolla, CA). Differences with a P⩽0.05 were considered significant.
References
Dye, C., Glaziou, P., Floyd, K. & Raviglione, M. Prospects for tuberculosis elimination. Annu. Rev. Public Health 34, 271–286 (2013).
Bifani, P.J., Mathema, B., Kurepina, N.E. & Kreiswirth, B.N. Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains. Trends Microbiol. 10, 45–52 (2002).
Kremer, K. et al. Definition of the Beijing/W lineage of Mycobacterium tuberculosis on the basis of genetic markers. J. Clin. Microbiol. 42, 4040–4049 (2004).
Kruuner, A. et al. Spread of drug-resistant pulmonary tuberculosis in Estonia. J. Clin. Microbiol. 39, 3339–3345 (2001).
Glynn, J.R., Whiteley, J., Bifani, P.J., Kremer, K. & van Soolingen, D. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis 8, 843–849 (2002).
Caws, M. et al. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 4, e1000034 (2008).
Manca, C. et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl Acad. Sci. USA 98, 5752–5757 (2001).
Ordway, D. et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 179, 522–531 (2007).
Cooper, A.M. & Khader, S.A. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol. Rev. 226, 191–204 (2008).
Manca, C. et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon. Cytokine Res. 25, 694–701 (2005).
Kolls, J.K. & Khader, S.A. The role of Th17 cytokines in primary mucosal immunity. Cytokine Growth Factor Rev. 21, 443–448 (2010).
Khader, S.A. et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175, 788–795 (2005).
Khader, S.A. et al. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J. Immunol. 187, 5402–5407 (2011).
Gopal, R. et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 10, e1004099 (2014).
Dudakov, J.A., Hanash, A.M. & van den Brink, M.R. Interleukin-22: immunobiology and pathology. Annu. Rev. Immunol. 33, 747–785 (2015).
Aujla, S.J. et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14, 275–281 (2008).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Dhiman, R. et al. Interleukin 22 inhibits intracellular growth of Mycobacterium tuberculosis by enhancing calgranulin A expression. J. Infect. Dis. 209, 578–587 (2014).
Whittington, H.A., Armstrong, L., Uppington, K.M. & Millar, A.B. Interleukin-22: a potential immunomodulatory molecule in the lung. Am. J. Respir. Cell Mol. Biol. 31, 220–226 (2004).
Sonnenberg, G.F. et al. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 207, 1293–1305 (2010).
Kumar, P., Thakar, M.S., Ouyang, W. & Malarkannan, S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 6, 69–82 (2013).
Pociask, D.A. et al. IL-22 is essential for lung epithelial repair following influenza infection. Am. J. Pathol. 182, 1286–1296 (2013).
Scriba, T.J. et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J. Immunol. 180, 1962–1970 (2008).
Matthews, K. et al. Predominance of interleukin-22 over interleukin-17 at the site of disease in human tuberculosis. Tuberculosis (Edinb) 91, 587–593 (2011).
Tadokera, R. et al. Role of the interleukin 10 family of cytokines in patients with immune reconstitution inflammatory syndrome associated with HIV infection and tuberculosis. J. Infect. Dis. 207, 1148–1156 (2013).
Cowan, J. et al. Comparison of interferon-gamma-, interleukin (IL)-17- and IL-22-expressing CD4 T cells, IL-22-expressing granulocytes and proinflammatory cytokines during latent and active tuberculosis infection. Clin. Exp. Immunol. 167, 317–329 (2012).
Zhang, M. et al. Anti-tuberculosis treatment enhances the production of IL-22 through reducing the frequencies of regulatory B cell. Tuberculosis (Edinb) 94, 238–244 (2014).
Zhang, G. et al. An SNP selection strategy identified IL-22 associating with susceptibility to tuberculosis in Chinese. Sci. Rep. 1, 20 (2011).
Dhiman, R. et al. IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J. Immunol. 183, 6639–6645 (2009).
Zeng, G. et al. Membrane-bound IL-22 after de novo production in tuberculosis and anti-Mycobacterium tuberculosis effector function of IL-22+ CD4+ T cells. J. Immunol. 187, 190–199 (2011).
Behrends, J., Renauld, J.C., Ehlers, S. & Holscher, C. IL-22 is mainly produced by IFNgamma-secreting cells but is dispensable for host protection against Mycobacterium tuberculosis infection. PLoS ONE 8, e57379 (2013).
Wilson, M.S. et al. Redundant and pathogenic roles for IL-22 in Mycobacterial, Protozoan, and Helminth Infections. J. Immunol. 184, 4378–4390 (2010).
Manca, C. et al. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J. Immunol. 162, 6740–6746 (1999).
Monin, L. & Khader, S.A. Chemokines in tuberculosis: the good, the bad and the ugly. Semin. Immunol. 26, 552–558 (2014).
Reed, M.B. et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87 (2004).
Ashiru, O.T., Pillay, M. & Sturm, A.W. Adhesion to and invasion of pulmonary epithelial cells by the F15/LAM4/KZN and Beijing strains of Mycobacterium tuberculosis. J. Med. Microbiol. 59, 528–533 (2010).
Yao, S. et al. Differentiation, distribution and gammadelta T cell-driven regulation of IL-22-producing T cells in tuberculosis. PLoS Pathog. 6, e1000789 (2010).
Dhiman, R. et al. NK1.1+ cells and IL-22 regulate vaccine-induced protective immunity against challenge with Mycobacterium tuberculosis. J. Immunol. 189, 897–905 (2012).
Mehra, S. et al. Reactivation of latent tuberculosis in rhesus macaques by co-infection with Simian Immunodeficiency Virus. J. Med. Primatol. 40, 485–494 (2011).
Slight, S.R. et al. CXCR5+ T helper cells mediate protective immunity against tuberculosis. J. Clin. Invest. 123, 712–726 (2013).
Choi, S.M. et al. Innate Stat3-mediated induction of the antimicrobial protein Reg3gamma is required for host defense against MRSA pneumonia. J. Exp. Med. 210, 551–561 (2013).
Desvignes, L., Wolf, A.J. & Ernst, J.D. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J. Immunol. 188, 6205–6215 (2012).
Gopal, R. et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am. J. Respir. Crit. Care Med. 188, 1137–1146 (2013).
Acknowledgements
This work was supported by Washington University in St Louis, NIH grants HL105427 to S.A.K., and AI111914 to S.A.K. and D.K., Alexander and Gertrude Berg Fellowship from the Department of Molecular Microbiology, Washington University St Louis to J.T. Funding and training support for O.P. was provided by Washington University Division of Pulmonary and Critical Care Medicine training grant “Principles in Pulmonary Research”, NIH/NHLBI T32 HL007317-38. J.R.-M. was supported by funds of the Department of Medicine, University of Rochester. We thank Sarah Squires for animal breeding, and Drs Kristin Griffiths, Mushtaq Ahmed for technical assistance with flow cytometry and BSL-3 experiments. We thank Dr Wenjun Ouyang, while at Genentech for the Il22−/− mice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Author contributions
P.T., O.P., J.R.-M., J.K.K. and S.A.K designed the experiments; P.T., O.P., A.C-L., M.M-T., M.A.S-L., B.F-J, D.K. and J.R-M did the experiments; M.S. and J.Z., provided human samples; P.T., OP and S.A.K., wrote the paper; D.K., T.R., J.Z., J.K.K., J.F.A. and S.A.K edited the paper, and S.A.K., and D.K. provided the funding.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
About this article

Cite this article
Treerat, P., Prince, O., Cruz-Lagunas, A. et al. Novel role for IL-22 in protection during chronic Mycobacterium tuberculosis HN878 infection. Mucosal Immunol 10, 1069–1081 (2017). https://doi.org/10.1038/mi.2017.15
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.15
This article is cited by
-
Helper T cell bias following tuberculosis chemotherapy identifies opportunities for therapeutic vaccination to prevent relapse
npj Vaccines (2023)
-
Role of tertiary lymphoid organs in the regulation of immune responses in the periphery
Cellular and Molecular Life Sciences (2022)
-
T-helper 22 cells develop as a distinct lineage from Th17 cells during bacterial infection and phenotypic stability is regulated by T-bet
Mucosal Immunology (2021)
-
Multidimensional analyses reveal modulation of adaptive and innate immune subsets by tuberculosis vaccines
Communications Biology (2020)
-
Th1 cytokines, true functional signatures for protective immunity against TB?
Cellular & Molecular Immunology (2018)

{kind=link}
{kind=link}
{kind=link}