
Abstract
Novel tuberculosis (TB)-vaccines preferably should (i) boost host immune responses induced by previous BCG vaccination and (ii) be directed against Mycobacterium tuberculosis (Mtb) proteins expressed throughout the Mtb infection-cycle. Human Mtb antigen-discovery screens identified antigens encoded by Mtb-genes highly expressed during in vivo murine infection (IVE-TB antigens). To translate these findings towards animal models, we determined which IVE-TB-antigens are recognised by T-cells following Mtb challenge or BCG vaccination in three different mouse strains. Eleven Mtb-antigens were recognised across TB-resistant and susceptible mice. Confirming previous human data, several Mtb-antigens induced cytokines other than IFN-γ. Pulmonary cells from susceptible C3HeB/FeJ mice produced less TNF-α, agreeing with the TB-susceptibility phenotype. In addition, responses to several antigens were induced by BCG in C3HeB/FeJ mice, offering potential for boosting. Thus, recognition of promising Mtb-antigens identified in humans validates across multiple mouse TB-infection models with widely differing TB-susceptibilities. This offers translational tools to evaluate IVE-TB-antigens as diagnostic and vaccine antigens.
Similar content being viewed by others

Introduction
Tuberculosis (TB) remains the most widespread and deadly infectious disease from a single bacterial pathogen worldwide1. The transmission of its causative agent, Mycobacterium tuberculosis (Mtb), cannot be sufficiently controlled -in adolescents and adults- by childhood vaccination with Bacillus Calmette-Guérin (BCG), the only currently licensed TB vaccine2. Therefore, numerous efforts in the last decades have attempted to develop better TB vaccines3. Two recent and independent clinical efficacy trials showed that BCG re-vaccination led to a decrease in sustained Mtb infection, and that multiple injections of an adjuvanted TB subunit vaccine, M72/AS01E, were able to prevent development of TB in latently Mtb infected individuals4,5. Although the efficacy of M72/AS01E still needs to be corroborated in larger cohorts and across different geographical areas6, this finding underscores the clear potential of using selected Mtb antigens as BCG booster vaccines2. However, immune correlates of TB protection remain unknown, posing a challenge to the identification of such protective Mtb antigens for vaccine design7.
In recent years, we have identified a class of Mtb antigens, named IVE-TB antigens, encoded by Mtb genes that are highly and consistently expressed in the lung of TB susceptible (C3HeB/FeJ) as well as TB resistant (C57BL/6J) mice following aerosol Mtb (Erdman) challenge8,9. Because these Mtb genes are highly expressed in vivo, the encoded IVE-TB proteins represent an interesting set of candidate antigens for several reasons. First of all, they are preserved among at least 219 Mtb clinical isolates and other pathogenic mycobacteria, thus offering the potential to elicit immune responses against multiple human virulent Mtb strains. Secondly, they share high homology with BCG and are thus likely implementable in booster vaccine strategies. Additionally, they contain multiple predicted HLA-Ia and HLA-II binding motifs covering 85% of the human population. Lastly but most importantly, many IVE-TB antigens were found to be recognised by immune cells of latently Mtb infected (LTBI) subjects and TB patients9,10. Interestingly, these T cells were capable of producing multiple cytokines, in addition to or even in the absence of IFN-γ, a cytokine known to be necessary but not sufficient in conferring protection against TB9,10.
Due to the absence of human Mtb challenge models and validated correlates of protection, promising candidate vaccines are commonly tested in mice before being evaluated in larger animal models and eventually clinical trials11. Many Mtb antigens, such as those included in the most advanced candidates in the TB vaccine pipeline12,13,14,15,16, have been screened previously in C57BL/6 and BALB/c17. Both strains (particularly C57BL/6) are relatively resistant to TB and do not display the full spectrum of human TB pathology, such as the development of centrally necrotic lesions18. By contrast, the less frequently used C3HeB/FeJ strain (also known as the Kramnik model) presents with necrotic lesions and lung cavitation resembling those found in human TB19.
In order to translate our previous findings from human cohorts towards highly controlled experimental in vivo TB models in which protective efficacy of candidate vaccines can be explored, we aimed to: (I) evaluate promising IVE-TB antigens across different strains of Mtb infected mice, including not only TB resistant (C57BL/6, BALB/c), but also TB susceptible (C3HeB/FeJ) strains, reasoning that the recognition of antigens in mice with different genetic backgrounds, and widely different TB susceptibilities, would better reflect the TB heterogeneity found in the human population; (II) study whether IVE-TB antigens induce IFN-γ as well as non-IFN-γ cytokine responses (TNF-α and IL-17) also in mice and in different organs (i.e., spleen, lung and mediastinal draining lymph nodes), to enable identification of those antigens that, though recognised in all mouse strains, would induce different cytokines profiles in resistant and susceptible mice. Upon asserting that an antigen is recognised in the susceptible strain, the cytokine and cellular responses can then be redirected towards a protective response (e.g., the response found in the resistant strains) by vaccination using appropriate adjuvants. (III) To determine which IVE-TB antigens recognised during Mtb infection are also recognised following vaccination with BCG, with the rationale to discriminate between highly specific Mtb antigens that could be useful candidates for TB diagnostics in areas with standard neonatal BCG vaccination programs, versus Mtb/BCG shared antigens that can be used as BCG booster vaccines; (IV) and validate findings in a multi-laboratory approach across two different laboratories and experimental settings to strengthen confidence in the results, especially in light of the large heterogeneity distinctive of human TB. The latter conforms to recent recommendations, which encourage researchers to perform pre-clinical discovery screenings in different mouse strains, routes of infection, Mtb strains and laboratories, before moving to more costly and resource demanding animal and human studies17.
We find 19 Mtb antigens that are significantly recognised by cells isolated from different murine tissues, presenting a wide array of immune responses with respect to the cytokines produced and the three mouse genetic backgrounds investigated. Of note, the recognition of 11 of these antigens was corroborated in an independent setting. Thus, our multi-laboratory approach identified promising Mtb antigenic targets for potential application as tools in TB control.
Results
IVE-TB antigen recognition in multiple organs of three genetically different mouse strains by cells producing different cytokines
Promising TB candidate vaccine antigens first need to be validated in small animal models before they can be selected for evaluation in larger animal models such as non-human primates, and eventually for testing in human clinical trials11. Here, we aimed to verify which IVE-TB antigens, previously shown to be recognised by T cells from TB patients and LTBI subjects9,10, were consistently and broadly recognised during the course of murine Mtb infection in different genetic backgrounds, given the fact that human populations are genetically highly diverse. To include different genetic backgrounds with widely diverse TB-susceptibility phenotypes, we selected C57BL/6, BALB/c and C3HeB/FeJ mice and challenged these with Mtb20,21. Different cell pools from each organ (spleens, lungs and mediastinal lymph nodes (medLN)), strain and time-points (early (5 weeks) or later (9–12 weeks) stage of Mtb infection) were tested in parallel by stimulation with single or fusion IVE-TB antigens (n = 22) (Table 1) as well as with positive controls (ConA and PWM) (Fig. 1a, Supplementary Data 1). Besides IVE-TB antigens, also several other, stage specific- and secreted-Mtb antigens (n = 5) were included in the screening since several studies previously described these as promising (diagnostic) TB biomarkers or as putative antigens for TB vaccination22,23,24,25 (Table 1). The concentrations of IFN-γ, TNF-α and IL-17 were measured in the cell supernatants after 3 days of stimulation. Cytokine levels in response to each Mtb antigen were compared between naïve mice and Mtb challenged mice for each organ and mouse strain, after subtracting background concentrations from unstimulated negative control samples (Supplementary Data 1).

a C57BL/6 (BL6), BALB/c (BALB) and C3HeB/FeJ (C3H) mice were challenged with 1 × 104 CFU Mtb-lux strain intratracheally and killed at an early (5 weeks) or later (9–12 weeks) stage of Mtb infection. Uninfected naïve control mice were killed and tested in parallel. Pooled cells from lungs, mediastinal lymph nodes (medLN) or spleens were stimulated with single or fusion Mtb antigens or with positive control stimuli ConA and PWM. The concentrations of IFN-γ, TNF-α and IL-17 were measured in the cell supernatants after 3 days of stimulation and corrected for background. b Heatmaps of the specific Mtb antigen responses for each cytokine are shown. Colour gradients indicate the cytokine concentration, while asterisks indicate significant differences between the naïve and the Mtb challenged groups. White *p-value < 0.05, computed with Kruskal–Wallis test; black asterisk marks differences that remained significant after multiple test correction (q-value < 0.1, i.e., FDR-adjusted p-values of Mann–Whitney.test). c 3D plot of the Variable Importance in Projection (VIP) scores obtained from each PLS-DA model computed on the basis of the following known classes: (i) time points (i.e., naïve mice vs. mice killed at early or late point after Mtb infection); (ii) mouse strains (i.e., C57BL/6 (BL6), BALB/c (BALB) and C3HeB/FeJ (C3H) mice); (iii) organs (i.e., lungs, mediastinal lymph nodes or spleens); (iv) cytokines (i.e., IFN-γ, TNF-α and IL-17). Antigens with a VIP score >1 were considered to have an above average influence on the model and are depicted by solid dots. Antigens with a VIP score >1 for the cytokine PLS-DA model are depicted by solid red dots. Antigens with a VIP score <1 are depicted by empty dots and their names are omitted in the 3D plot.
Several trends emerged when grouping the cytokine responses against the 27 Mtb antigens included in this study (Supplementary Fig. 1). High TNF-α responses were found to the majority of the selected Mtb antigens, most predominantly in the spleen, in all three mouse strains. Secondly, the cells analysed from the C3HeB/FeJ mice showed an overall reduced ability to produce TNF-α against Mtb antigens, especially cells from the medLN and lungs whose TNF-α response was low or absent even after mitogen stimulation (Fig. 1b and Supplementary Fig. 1). Antigen-specific IFN-γ and IL-17 responses were more diversely distributed and varied strongly between organs and mouse strains (Fig. 1b). Interestingly, especially in BALB/c and C57BL/6 mice, significant IL-17 responses were mainly induced in cells isolated from the lungs and medLN, while IL-17 responses were almost absent among the splenocytes (Supplementary Fig. 1). For splenocytes and medLN, the highest peak of cytokine production was mostly observed at an early stage of Mtb infection, while for lung cells this was observed at the later stage (Supplementary Fig. 1). IFN-γ responses tended to be highest in C3HeB/FeJ mice, the most TB susceptible strain, confirming that like in humans IFN-γ in itself is not sufficient for protective immunity.
Among the Mtb antigens that induced a significant specific increase in cytokine production, the highest number was recognised by medLN cells from C57BL/6 mice (18/27 antigens recognised), followed by C3HeB/FeJ (7/27 antigens recognised) and BALB/c (4/27 antigens recognised) mice (Fig. 1b).
From these antigens, only one was recognised by all mouse strains (Rv0826), five by C57BL/6 and C3HeB/FeJ (Rv1886, Rv0440, Rv1980, Rv1791 and Rv1733), and 3 by C57BL/6 and BALB/c mice (Rv1131, Rv1221 and Rv0470). In the C57BL/6 mice and, to a lesser extent, in the other two mouse strains, medLN cells produced multiple cytokines in response to single Mtb antigens. The different combinations found were: IFN-γ, TNF-α and IL-17 against 3 antigens (in C57BL/6 mice: Rv1886, Rv1131 and Rv1980); IFN-γ and TNF-α against 5 antigens (in C57BL/6 mice: Rv0287/88, Rv0470, Rv1791, ESAT6/CFP10 and Rv0440. For Rv0440, the same cytokine profile was found in C3HeB/FeJ mice); IFN-γ and IL-17 against two antigens (in C3HeB/FeJ mice: Rv0826 and Rv1980); TNF-α and IL-17 against 2 antigens (in C57BL/6 mice: Rv3616; in BALB/c mice: Rv1131).
Antigen-specific splenocyte responses to Mtb antigens were mainly detected – as measured by either one or more of the three cytokines evaluated – in C57BL/6 mice (11 antigens recognised) and to a lesser extent in BALB/c (3 antigens recognised) and C3HeB/FeJ (2 antigens recognised) mice. Among these, only the recognition of one antigen, Rv1886, was shared between C57BL/6 and BALB/c mice, and one other (Rv1791) between C57BL/6 and C3HeB/FeJ. Only splenocytes from C57BL/6 mice produced multiple cytokines, i.e., IFN-γ and TNF-α, against 5 antigens (Rv1886, Rv0287/88, Rv0470, Rv1791 and Rv3615).
Lung cells consistently recognised fewer antigens than cells isolated from other tissues (Fig. 1c). However, some of these antigens were recognised by lung cells of different mouse strains (Rv0287/88 was recognised by all mouse strains; Rv1886 by C57BL/6 and BALB/c; ESAT6/CFP10 and Rv1791 by C57BL/6 and C3HeB/FeJ). Significant levels of IFN-γ and IL-17 were secreted by lung cells in response to 4 antigens (in C57BL/6 mice: Rv0287/88 and Rv0470; in BALB/c mice: Rv1886; and in C3HeB/FeJ mice: Rv1791). Only against one antigen (Rv0287/88) a combined IFN-γ, TNF-α and IL-17 response was found in BALB/c mice.
Most of the differences listed above were no longer significant after the very stringent FDR correction, but importantly this included also responses to controls and to previously described antigens known to be highly immunogenic such as ESAT6/CFP10 (Figs 1b and 2c). Therefore, the results are shown both with and without FDR correction, to avoid the risk of missing biological relevant candidates at this early stage of discovery.

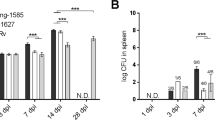
a C3HeB/FeJ mice were either challenged intranasally with H37Rv-Mtb strain (105 CFU) (10 mice), vaccinated subcutaneously with BCG (5 mice) or left untreated (5 mice). After 6 weeks, splenocytes were harvested and stimulated per mouse for 6 days with a set of eighteen Mtb antigens or positive controls (ConA, PPD, bead disrupted BCG (BCG bub) and Mtb lysate), or negative control (HPV16E6 recombinant protein). The IFN-γ concentrations detected in the unstimulated samples were subtracted from the stimulated samples for each condition within each mouse. b IFN-γ production after antigen stimulation in splenocytes from naïve unimmunized mice (left panel), and BCG immunised versus Mtb infected mice (right panel). Dots display the median IFN-γ production. The (Rv) codes of the antigens are indicated if the median IFN-γ production exceeded 500 pg/ml in both BCG immunised and Mtb infected mice. The horizontal dotted line indicates the cut-off at 500 pg/ml, which is twice the median IFN-γ production found in response to the negative control in BCG immunised and Mtb infected mice (right panel). c IFN-γ responses against Mtb antigens in splenocytes from naïve, BCG immunised and Mtb infected mice are shown. Dots represent a single mouse (10 mice in the Mtb infected group and 5 mice in the naïve and BCG immunised group in total) and bars indicate the median IFN-γ response of each group. The Mtb challenged and BCG immunised groups were compared to the naïve group using the Kruskal–Wallis test and a p-value < 0.05 was considered significant. Asterisk marks differences that remained significant after multiple test correction (q-value < 0.1, i.e., FDR-adjusted p-values of Mann–Whitney test).
Interestingly, in the resistant mouse strains, there were a few antigens (Rv0470, Rv0826, Rv0991, Rv1131, Rv1221, Rv1733, Rv1846, Rv1980, Rv2034, Rv2873, Rv3052, Rv3353 and Rv3616) that induced statistically significant TNF-α but not IFN-γ responses. Except for Rv1846, this lack of overall significant IFN-γ responses seemed to be the result of the observed heterogeneity in donor responses rather than the complete absence of this cytokine (Supplementary Fig. 3).
To validate the results in a subsequent independent multivariate analytical approach, we used PLS-DA modelling to determine the key antigens that drove the discrimination in cytokine responses between (i) time points (E/C, Rv0642, Rv0287/88, Rv0991, Rv1791, Rv1886, Rv2029 and Rv3616); (II) mouse strains (E/C, Rv0287/88, Rv0440, Rv0470, Rv0642, Rv1221, Rv1791, Rv1886, Rv2029, Rv2873 and Rv3615); (iii) organs (E/C, Rv0287/88, Rv1131, Rv1791, Rv1886, Rv2029, Rv3048, Rv3615 and Rv3616); and (iv) type of cytokine (E/C, Rv0287/88, Rv0440, Rv1131, Rv1791, Rv1886, Rv1980, Rv2029 and Rv3615), based on the Variable Importance in Projection (VIP) values (VIP > 1). The PLS-DA protein clusters (Supplementary Fig. 2a) were very similar to those obtained by unsupervised PCA (Supplementary Fig. 2b) confirming their validity. (Supplementary Data 1 and Fig. 1c). Of note, this independent multivariate approach confirmed 13 of the 19 proteins that were found as being interesting targets in our first analysis.
Overall, nineteen out of 27 selected Mtb antigens (Fig. 1c, in bold) elicited significant cytokine production from cells isolated from Mtb infected mice compared to those found in uninfected animals. Of note, although as expected these responses were somewhat heterogeneous, 13 antigens appeared to drive discriminatory cytokine responses between the different mouse strains, time points, type of cytokine and tissues analysed in this study. But perhaps most importantly, the results show that human T cell recognition profiles translate well to mouse models, which was a major aim of this study.
IVE-TB antigen-specific responses after live BCG vaccination
A TB subunit vaccine, in order to be effective, should preferably boost immune responses induced by prior vaccination26. BCG, despite its insufficient protective efficacy against adult TB, remains the only licensed vaccine against TB and is highly effective in protecting against severe childhood TB2. To evaluate whether immune responses against IVE-TB antigens could be induced by BCG immunisation, and to compare those responses to those induced by Mtb infection, C3HeB/FeJ mice were either challenged with H37Rv-Mtb strain (105 CFU) (10 mice), vaccinated subcutaneously with BCG (5 mice) or left untreated (5 mice) (Fig. 2a). After 6 weeks, splenocytes were harvested and stimulated for 6 days with a set of 18 Mtb antigens selected from our first screen (Fig. 1c). Positive (ConA, PPD, bead disrupted BCG, Mtb lysate) and negative (HPV16E6 recombinant protein) controls were also included. The IFN-γ production in response to the different stimuli was measured and analysed, and as above concentrations detected in the unstimulated samples were subtracted from each condition within each mouse. TNF-α and IL-17 responses were only present at low levels in C3H mice in the first experiment (Fig. 1b) and therefore not assessed in this screening. Three antigens (Rv0470, Rv1733 and Rv3616) were strongly recognised by Mtb infected as well as BCG vaccinated C3HeB/FeJ mice (i.e.: IFN-γ production >500 pg/ml) (Fig. 2b). Additionally, despite the different experimental setup and laboratory setting, eleven Mtb antigens (Rv0287/88, Rv1886, Rv0470, Rv1131, Rv0440, Rv1980, ESAT6/CFP10, Rv3616, Rv1733, Rv1221 and Rv3052) were confirmed to be significantly recognised after Mtb challenge in C3HeB/FeJ mice (Fig. 2c). Interestingly, besides ESAT6/CFP10 (E/C), of which peptides are already widely used as TB diagnostic tool in IGRA tests27, Rv0287/88, Rv1886, Rv1131, Rv1221, Rv1846 and Rv3052 were the only antigens for which IFN-γ responses differed highly between the Mtb infected and the BCG vaccinated group (Fig. 2c). Prior to FDR correction, differences between the Mtb infected and the BCG vaccinated group were significant only for E/C (as expected), Rv3052 and Mtb lysate.
Collectively, these findings confirm, extend and partially validate the immune recognition of eleven Mtb antigens previously identified in humans across three genetically unrelated mouse TB-infection models and two different independent laboratory settings.
Discussion
An important requirement for novel TB candidate vaccines is that they evoke immunity to Mtb proteins that are expressed, processed and presented to the host immune system during infection. Over the last years, we identified multiple Mtb proteins recognised by immune blood cells from latently Mtb infected individuals and TB patients3,9. During the antigen selection process, we selected proteins encoded by genes highly expressed either during the entire course of infection, or during specific stages of in vivo Mtb infection3. Before entering clinical studies, candidate vaccines need to be evaluated and proven to be immunogenic in animal studies11. Therefore, we here examined a selection of Mtb antigens identified in our previous in vitro human studies and backtranslated this to three different, genetically and TB-susceptibility diverse mouse TB models. We found that among a set of 27 Mtb antigens recognised by human cells, 19 were able to induce the production of at least one of three cytokines (IFN-γ, TNF-α, IL-17) in at least one of the tissues examined in each of the three mouse strains after challenge with Mtb. Further confirmation of our previous data in human cohorts was that several Mtb antigens induced considerable cytokine responses other than IFN-γ9,28. Importantly, and in accordance with current recommendations17, the results were partially validated in a second independent laboratory and experimental setting (Supplementary Table 1), showing recognition of 11 Mtb antigens in infected C3HeB/FeJ mice. Among those, three antigens (i.e.: Rv0470, Rv1733 and Rv3616) were found to be recognised also after BCG vaccination. The latter finding is essential when considering the design of BCG-booster vaccines against TB2.
Overall, in the experiments performed in the first laboratory, we observed highly significant responses to Mtb antigens which varied by magnitude, cytokine combinatorial patterns, tissues and mouse strains (Fig. 1). As previously observed in human studies9, the differences in the levels and types of cytokines induced by an antigen, or the fact that certain antigens did not elicit detectable immune responses, is not linked to a particular protein function. Interestingly, non-IFN-γ responses were detected in the spleen and the mediastinal draining lymph nodes of the resistant C57BL/6 mouse strain after stimulation with a substantial number of antigens (Rv0470, Rv0826, Rv0991, Rv1131, Rv1221, Rv1733, Rv1846, Rv1980, Rv2034, Rv2873, Rv3353 and Rv3616), which agrees with our previous findings9 and also with the recent description of non-canonical IFN-γ-independent immune responses found in the peripheral blood of Mtb exposed “resistant” individuals28. Cells from C57BL/6 mice recognised significantly more Mtb antigens than those from BALB/c and C3HeB/FeJ mice. Although C57BL/6 and BALB/c mice are equally resistant to TB29, several genetic determinants might have contributed to the differences found in the antigens recognised. For instance, the adaptive type 1 immune response to pulmonary mycobacterial infection appears later and with lower magnitude in BALB/c compared to C57BL/6 mice30,31. In line with our findings, the lower magnitude of IFN-γ and TNF-α production levels in BALB/c mice was already described in splenocytes and lung lymph node cells upon antigen stimulation31. Unexpectedly to us, we found that lung and mediastinal lymph nodes cells from C3HeB/FeJ mice failed to produce detectable TNF-α, not only after Mtb antigens exposure, but also upon some of the positive mitogen controls. During mycobacterial infection, TNF-α deficient mice develop necrotic granulomas, pathogenic lesions which are also characteristic of the C3HeB/FeJ mice32,33. Although cells isolated from lung of C3HeB/FeJ mice can produce TNF-α, they do that to a lesser extent than C57BL/6 mice, potentially explaining why they develop necrotic lesions. The lower TNF-α production in C3HeB/FeJ mice might be caused by a greater and premature accumulation of a functionally exhausted T cell subsets, characterised by a limited capacity to secrete IL-2 and TNF-α34. Demonstrating the recognition of Mtb antigens in this TB susceptible strain was particularly important from a corrective vaccine perspective. Redirecting the cellular response in such strains towards a protective response (e.g., that found in the resistant strain) could be achieved by immunising mice with recognised antigens and appropriate adjuvants (e.g. CAF01), as has been done for instance with ESAT6, which is a strongly antigenic protein in active TB patients, but is also a powerful antigen for vaccination when delivered in CAF0135.
Only two antigens were consistently recognised by cells isolated from the same tissue across all mouse strains: Rv0287/88 and Rv0826. Rv0287/88 is a heterodimer secreted by the ESX-3 secretion system, implicated in Mtb virulence and survival36,37. Lung cells from all three mouse models produced IFN-γ when stimulated with Rv0287/88, while TNF-α and IL-17 were secreted concomitantly in BALB/c mice and TNF-α in C57BL/6 mice. In the latter strain, also splenocytes and mediastinal lymph nodes cells responded to Rv0287/88 by producing IFN-γ and TNF-α. These findings validate and extend what has been shown for lung T cells of CB6F1 (BALB/c × C57BL/6) and B6C3F1 (C57BL/6 × C3HeB/FeJ), which are able to recognise Rv0287/88 after 21 days of Mtb infection38. Although evidence for the antigenicity of Rv0287/88 accumulates, susceptible C3HeB/FeJ mice immunised with BCG did not recognise this fusion protein. This contrasts with another study, performed in a TB-resistant strain, wherein splenocytes of BCG vaccinated mice responded to specific Rv0288 epitopes39. Rv0288 constitutes part of the H4:IC31, an adjuvanted polyprotein vaccine that, though immunogenic, failed to boost neonatal BCG vaccination in a recent prevention of (sustained) infection clinical trial4. Testing this vaccine in BCG immunised mice with different TB susceptibility phenotypes might provide improved models that are more predictive of humans.
At an early stage of Mtb infection, Rv0826 was recognised by medLN cells from all mice, mainly inducing TNF-α in C57BL/6 and BALB/c mice and IFN-γ and IL-17 in C3HeB/FeJ mice. Rv0826 is a conserved protein with unknown function, highly expressed during enduring hypoxia and in extensively drug resistant Mtb strains40,41. These characteristics, together with the strong cellular recognition found in humans42 and in mice, would make this antigen an attractive target. However, we observed consistent unspecific immune responses against Rv0826 in the splenocytes of all mice, verified in an independent experiment (Fig. 2b), which deserves further investigation. This might be due to cross-reactivity to other microbial antigens, e.g., microbiota derived, but this remains currently speculative.
Besides Rv0287/88 and Rv0826, other antigens were consistently recognised across tissues and mouse strains, and better able to elicit cells producing multiple cytokines (Rv1886, Rv1791, Rv0470, Rv1131, Rv0440, Rv1980) than other antigens (ESAT6/CFP10, Rv3615, Rv3616, Rv1733, Rv1221, Rv1846, Rv0991, Rv2034, Rv2873, Rv3052 and Rv3353). Among those, Rv0470c was particularly interesting because it is an IVE-TB antigen consistently recognised across mice both after Mtb challenge and after BCG vaccination, in independent experiments and in different laboratories. Additionally, though recognised in susceptible mice, Rv0470 induced different magnitudes and type of cytokine responses in the resistant strains (Fig. 1b, c). Rv0470c is an enzyme essential for both the synthesis and cyclopropanation of mycolic acids and the cording morphology of both BCG and Mtb43,44. In addition, Rv0470c prevents phagosome maturation in human monocyte-derived macrophages, while in mice, its deletion alters the persistence and pathology of Mtb at late stages of infection43,45. Of note, immunisation with BCG_0470 increased the delayed-type hypersensitivity as well as Th1 responses in BCG primed BALB/c mice46. All these features, supported by our data in mice and humans9,10, point to Rv0470c as an attractive BCG booster vaccine candidate when delivered with a properly selected adjuvant47.
Interestingly, Rv0287/88, Rv1886, Rv1131, Rv1221, Rv1846 and Rv3052, though highly homologous to M. bovis proteins, were the antigens, besides ESAT6/CFP10, for which IFN-γ response highly differed between Mtb infected and BCG vaccinated groups (Fig. 2c)9. Comparing the specific responses between TB patients and non Mtb exposed BCG vaccinees might reveal the potential of these antigens as adjunct TB diagnostic tools.
In conclusion, this study demonstrates the consistent recognition of several promising IVE-TB Mtb antigens among multiple genetically and TB susceptibility diverse Mtb infected mouse strains, across various infection models and tissues and time points evaluated, thus offering translational tools to evaluate Mtb antigens in appropriate animal models.
Methods
Intratracheal Mtb infection in C57BL/6, BALB/c and C3HeB/FeJ
C57BL/6, BALB/c and C3HeB/FeJ mice were bred in the animal facilities of Sciensano. Mice or breeding couples were obtained from Janvier Labs (France) for C57BL/6 and BALB/c or from The Jackson Laboratory (USA) for C3HeB/FeJ (JAX stock #000658). Aged-matched female mice 8–15 weeks old at the start of each experiment were used. All Mtb infections were performed in a biosafety level 3 (BSL3) facility at Sciensano and all the procedures were performed in accordance with the Belgian legislation and were approved by the ethics committee of Sciensano under the file number 201405–14–01.
Mice were infected intratracheally with a dose of 1 × 104 CFU of virulent and luminescent M. tuberculosis H37Rv grown for 2 weeks as a surface pellicle on Sauton medium and stored frozen in aliquots at −80 °C. The M. tuberculosis H37Rv strain used is transformed with the reporter plasmid pSMT1, which expresses the Vibrio harveyi luxAB genes under the control of the BCG hsp60 promoter21. We have previously shown that measuring RLU is an accurate alternative to determining CFU counts21,48. Bacterial loads in total lungs of infected mice were verified at the specified time-points in all infected mice for the first screen. For the second and third screens, bacterial loads were measured in the left lung lobe of individual mice of 1 pool/time-point. The other lobes and total lungs of the mice of the other pools were treated with Collagenase/DNAse I for isolation of leucocytes. To measure bacterial loads, the number of bioluminescent organisms [determined as relative light units (RLU)] in lung homogenates was determined by a bioluminescence assay with a Modulus luminometer (Turner Biosystems) and 1% n-decanal in ethanol as a substrate. Data are expressed as mean log10 mRLU values per group (Supplementary Data 1).
Preparation of cells harvested from intratracheal Mtb infected C57BL/6, BALB/c and C3HeB/FeJ mice
Uninfected naïve control mice and infected mice were killed at specified time-points (Supplementary Data 1). Spleens, lungs and mediastinal lymph nodes were removed aseptically and prepared for cell culture. For that purpose, spleens, mediastinal lymph nodes and lungs (first screen, Supplementary Data 1) were gently homogenised in RPMI medium supplemented with penicillin (60 µg/ml) using a Dounce homogenizer. Mediastinal lymph nodes and lungs (first screen, Supplementary Data 1) were passed through a 70-µm nylon cell strainer (Greiner). Lungs (second and third screen, Supplementary Data 1) were fragmented using a scalpel and digested for 1 h at 37 °C and 5% CO2 in HBSS (Gibco) supplemented with penicillin (60 µg/ml) and streptomycin (100 µg/ml), 5% of foetal calf serum (FCS), 1 mg/ml of Collagenase from Clostridium histolyticum (Sigma-Aldrich, C5138) and 0.1 mg/ml of DNAse I from bovine pancreas (Roche, 11284932001). After incubation, digested lung fragments were gently homogenised in a Dounce homogenizer and passed through a 70-µm nylon cell strainer (Greiner). Single cell solutions of cells isolated from spleen, mediastinal lymph nodes and lungs were washed, counted and cultured in RPMI medium supplemented with 5 × 10−5 M 2-mercaptoethanol, penicillin (60 µg/ml) and streptomycin (100 µg/ml), and 10% FCS (number of cells in Supplementary Data 1). Cells isolated from the same organ and mouse strain were pooled in order to have a sufficient number of leucocytes. Pools of cells isolated from 4–13 organs/experimental group were cultured for 72 h with recombinant proteins (10 µg/ml) or RPMI as a negative control (defined as unstimulated samples) or Pokeweed (PWM, 20 µg/ml, Sigma, Cat. N° L9379) or Concanavalin A (ConA, 4 µg/ml, Sigma, Cat. N° C5275) as positive controls. Cell free culture supernatants from at least three separate wells were pooled and supernatants were stored frozen at −20 °C until analysis by enzyme-linked immunosorbent assay (ELISA) was performed.
Intranasal Mtb infection and sub cutaneous BCG vaccination of C3HeB/FeJ
C3HeB/FeJ female mice purchased from the Jackson Laboratory (USA, JAX stock #000658) were used in this study at 6 weeks of age. C3HeB/FeJ mice were infected with 1 × 105 CFU live Mtb strain H37Rv from glycerol stocks, stored at −80 °C. Mice were anaesthetised with 2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane (isofluoran; Pharmachemie BV, The Netherlands) and intranasally (i.n.) infected. Six weeks post Mtb challenge, mice were euthanized with CO2 and lungs and splenocytes were aseptically removed. Organs were homogenised using 70 µM cell strainers (Corning, U.S.A.) and the amount of Mtb bacteria was determined by plating out serial dilutions of the homogenates on 7H11 plates (BD Bioscience, U.S.A), supplemented with BBL Middlebrook OADC enrichment (BD Bioscience, U.S.A) and PANTA (BD Bioscience, U.S.A). Colonies were counted after 3 weeks of incubation at 37 °C (Supplementary Data 2). For BCG immunisation, C3HeB/FeJ mice were injected subcutaneously in the right flank with 106 CFU BCG1331 (SSI, Denmark) from glycerol stocks, stored at −80 °C. Six weeks post BCG vaccination, mice were killed and splenocytes were aseptically removed. Naïve mice were included as control. All mice were daily monitored for ethical requirements, and weighed once a week according to the ethical regulations at the LUMC animal facility (DEC number 11183).
Recombinant proteins
A total of 27 Mtb recombinant proteins, previously identified by different Mtb antigen discovery approaches3, were tested in this study (Table 1). As described previously9, Mtb genes as well as the HPV16E6 gene, were amplified by PCR from genomic H37Rv DNA and cloned by Gateway technology (Invitrogen, Carlsbad, CA, USA) in a bacterial expression vector, overexpressed in Escherichia coli (E. coli) BL21 (DE3) and purified. Gel electrophoresis and western blotting with an anti-His tag Antibody (Invitrogen) and an anti-E. coli polyclonal antibody (a kind gift of Statens Serum Institute, Copenhagen, Denmark) were used to check the size and purity of the recombinant proteins. Rv0287/88 and Rv3874/75 were produced as fusion proteins to mirror the pairwise dependent secretion pathway by the mycobacterial T7S system. All recombinant proteins here included were previously tested to confirm lack of protein-non-specific T-cell stimulation and cellular toxicity9.
ELISA on cells harvested from intratracheally Mtb infected mice
IFN-γ was quantified by sandwich ELISA using purified rat anti-mouse IFN-γ clone R4-6A2 (BD Biosciences, Cat. No 551216) as the capture antibody, biotinylated rat anti-mouse IFN-γ clone XMG1.2 (BD Biosciences, Cat. No 554410) as detection antibody, peroxidase-conjugated streptavidin (Jackson ImmunoResearch, Cat. No 016-030-084) and recombinant mouse IFN-γ protein (R&D Systems, Cat. No 485-MI) as standard. Plates were revealed with O-phenylenediamine dihydrochloride substrate (SIGMAFAST™ OPD, Cat. No P9187), the reaction was stopped with 1 M H2SO4, and the optical density was read at 490 nm on an Infinite F50 plate-reader (Tecan).
TNF-α and IL-17A were quantified by sandwich ELISA with respectively TNF-α mouse uncoated ELISA kit (Invitrogen, Cat. No 88-7324-88) and IL-17A (homodimer) mouse uncoated ELISA kit (Invitrogen, Cat. No 88-7371-88) following the recommended manufacturer’s procedures and optical density was read at 450 nm on an Infinite F50 plate-reader (Tecan).
The respective dynamic ELISA assay ranges are 20–5000 pg/mL for IFN-γ; 8–1000 pg/mL for TNF-α and 4–500 pg/mL for IL-17A and appropriate serial dilutions of the samples were tested to quantify these three different cytokines.
ELISA results were analysed by converting measured absorbance to concentrations (pg/ml) by linear regression of the optical densities measured for the diluted standards using the Magellan for F50 software (Tecan).
ELISA on samples form intranasally Mtb infected and subcutaneously BCG vaccinated mice
Mtb infected splenocytes (300,000 cells/well) were incubated with single recombinant proteins or mitogens (5 µg/ml) at 37°, 5% CO2. After 6 days of stimulation, the supernatants were filtered through 0.2 µM filter plates (Corning, U.S.A.). The IFN-γ ELISA (BD Bioscience, U.S.A, capture antibody cat no 551216 and biotin labelled detection antibody cat no 554410) was performed on the filtered supernatants according to manufacturer’s instructions. Absorbance (OD450) was determined and converted to concentrations using a standard curve by the Microplate Manager software version 5.2.1 (Biorad Laboratories, The Netherlands) (Supplementary Data 2).
Data analysis
For each cytokine, within each pool of mice (Supplementary Data 1) or single mouse (Supplementary Data 2), the background-value, i.e., the value determined for unstimulated samples, was subtracted from the value determined for each antigen or mitogen. Data obtained at the later stage are pooled for analysis (9–12 weeks). The Kruskal–Wallis test was performed to assess the difference between the responses found in uninfected mice versus infected mice at different time points or versus BCG immunised mice for each antigen using IBM SPSS Statistics 26 (Figs 1b and 2b, Supplementary Data 1 and 2). Due to the high number of comparisons included in the study, we performed a two-sided Mann-Whitney test with an FDR (Benjamini-Hochberg) correction (results reported in the Supplementary Data 1 and 2). However, significant responses to positive controls or to previously described antigens (such as ESAT6/CFP10) were lost after correction (with an FDR cut-off set at 0.1), questioning whether this more stringent approach could lead to the exclusion of interesting candidates at this early stage of discovery. Thus, results of both unadjusted and adjusted tests are reported. In addition, for the first set of experiments, we analysed the responses to specific antigens on the basis of known classes ((i) naïve mice vs. mice killed at early or late point after Mtb infection; (II) mouse strains; (iii) organs; (iv) cytokines) using a supervised partial least squares-discriminant analysis (PLS-DA) method (Supplementary Fig. 2a) and a principal component analysis (PCA) (Supplementary Fig. 2b). The median responses to each specific antigen (Supplementary Data 1) were explored and analysed with ‘mixOmics’ (package version 6.15, http://www.mixOmics.org) in R (version 4.0.3) after log10 transformation. Three-component models explained the highest proportion of variance in the PCA as well as lowest balanced error rate (BER) in the PLSA-DA (Supplementary Data 1). For each of the categories investigated through PLSA-DA, we obtained Variable Importance in Projection (VIP) values which are a quantitative estimation of the discriminatory power of each individual antigen in a specific model (Supplementary Data 1 and Fig. 1c). The VIP values vary in a fixed range since the sum of squared VIP scores for all variables sum to the number of variables49. Therefore, antigens with a VIP value larger than 1 (i.e., larger than the average of squared VIP values) were considered to have an above average influence on the model49.
The Kruskal–Wallis test with Dunn’s multiple test correction was applied in Graphpad Prism (version 8) to analyse cytokine responses measured for all the 27 Mtb antigens across organs and timepoints (Supplementary Fig. 1).
Of note, in the second experiment, the IFN-γ response to HPV16E6, used as negative control, was higher in mice after BCG or Mtb infection than in the uninfected group. This response could be due to a non-specific immune activation profile. Therefore, we considered responses to be significant and specific only when the median IFN-γ production exceeded twice the IFN-γ production found in response to the negative control in BCG immunised and Mtb infected mice.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Code availability
The code used in this study was obtained from the ‘mixOmics’ (package version 6.15, http://www.mixOmics.org) for R (version 4.0.3).
References
WHO. World Health Organization: Global tuberculosis report 2019 (2019).
Schrager, L. K., Vekemens, J., Drager, N., Lewinsohn, D. M. & Olesen, O. F. The status of tuberculosis vaccine development. Lancet Infect. Dis. 20, e28–e37 (2020).
Coppola, M. & Ottenhoff, T. H. Genome wide approaches discover novel Mycobacterium tuberculosis antigens as correlates of infection, disease, immunity and targets for vaccination. Semin. Immunol. 39, 88–101 (2018).
Nemes, E. et al. Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N. Engl. J. Med. 379, 138–149 (2018).
Tait, D. R. et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N. Engl. J. Med. 381, 2429–2439 (2019).
Ottenhoff, T. H. M. A trial of M72/AS01E vaccine to prevent tuberculosis. N. Engl. J. Med. 382, 1576–1577 (2020).
Zhu, B., Dockrell, H. M., Ottenhoff, T. H. M., Evans, T. G. & Zhang, Y. Tuberculosis vaccines: opportunities and challenges. Respirology 23, 359–368 (2018).
Commandeur, S. et al. An unbiased genome-wide Mycobacterium tuberculosis gene expression approach to discover antigens targeted by human T cells expressed during pulmonary infection. J. Immunol. 190, 1659–1671 (2013).
Coppola, M. et al. New genome-wide algorithm identifies novel in-vivo expressed mycobacterium tuberculosis antigens inducing human T-cell responses with classical and unconventional cytokine profiles. Sci. Rep. 6, 37793 (2016).
Coppola, M. et al. Cell-mediated immune responses to in vivo-expressed and stage-specific mycobacterium tuberculosis antigens in latent and active tuberculosis across different age groups. Front. Immunol. 11, 103 (2020).
Kaufmann, S. H. E. et al. TBVAC2020: advancing tuberculosis vaccines from discovery to clinical development. Front. Immunol. 8, 1203 (2017).
Skeiky, Y. A. et al. Differential immune responses and protective efficacy induced by components of a tuberculosis polyprotein vaccine, Mtb72F, delivered as naked DNA or recombinant protein. J. Immunol. 172, 7618–7628 (2004).
Ryter, K. T. et al. Aryl trehalose derivatives as vaccine adjuvants for Mycobacterium tuberculosis. J. Med. Chem. 63, 309–320 (2020).
Brandt, L., Elhay, M., Rosenkrands, I., Lindblad, E. B. & Andersen, P. ESAT-6 subunit vaccination against Mycobacterium tuberculosis. Infect. Immun. 68, 791–795 (2000).
Aagaard, C. et al. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 17, 189–194 (2011).
Kwon, K. W. et al. Long-term protective efficacy with a BCG-prime ID93/GLA-SE boost regimen against the hyper-virulent Mycobacterium tuberculosis strain K in a mouse model. Sci. Rep. 9, 15560 (2019).
Franzblau, S. G. et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 92, 453–488 (2012).
Singh, A. K. & Gupta, U. D. Animal models of tuberculosis: Lesson learnt. Indian J. Med. Res. 147, 456–463 (2018).
Kramnik, I. & Beamer, G. Mouse models of human TB pathology: roles in the analysis of necrosis and the development of host-directed therapies. Semin. Immunopathol. 38, 221–237 (2016).
Romano, M. et al. Immunogenicity and protective efficacy of tuberculosis subunit vaccines expressing PPE44 (Rv2770c). Vaccine 26, 6053–6063 (2008).
Snewin, V. A. et al. Assessment of immunity to mycobacterial infection with luciferase reporter constructs. Infect. Immun. 67, 4586–4593 (1999).
van Pinxteren, L. A., Ravn, P., Agger, E. M., Pollock, J. & Andersen, P. Diagnosis of tuberculosis based on the two specific antigens ESAT-6 and CFP10. Clin. Diagn. Lab. Immunol. 7, 155–160 (2000).
Commandeur, S. et al. Double- and monofunctional CD4(+) and CD8(+) T-cell responses to Mycobacterium tuberculosis DosR antigens and peptides in long-term latently infected individuals. Eur. J. Immunol. 41, 2925–2936 (2011).
Serra-Vidal, M. M. et al. Immunogenicity of 60 novel latency-related antigens of Mycobacterium tuberculosis. Front. Microbiol. 5, 517 (2014).
Karbalaei Zadeh Babaki, M., Soleimanpour, S. & Rezaee, S. A. Antigen 85 complex as a powerful Mycobacterium tuberculosis immunogene: biology, immune-pathogenicity, applications in diagnosis, and vaccine design. Microb. Pathog. 112, 20–29 (2017).
Kaufmann, S. H. E. Vaccination against tuberculosis: revamping BCG by molecular genetics guided by immunology. Front. Immunol. 11, 316 (2020).
Zaheen, A. & Bloom, B. R. Tuberculosis in 2020 - new approaches to a continuing global health crisis. N. Engl. J. Med. 382, e26 (2020).
Lu, L. L. et al. IFN-gamma-independent immune markers of Mycobacterium tuberculosis exposure. Nat. Med. 25, 977–987 (2019).
Medina, E. & North, R. J. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology 93, 270–274 (1998).
Arko-Mensah, J. et al. Resistance to mycobacterial infection: a pattern of early immune responses leads to a better control of pulmonary infection in C57BL/6 compared with BALB/c mice. Vaccine 27, 7418–7427 (2009).
Wakeham, J., Wang, J. & Xing, Z. Genetically determined disparate innate and adaptive cell-mediated immune responses to pulmonary Mycobacterium bovis BCG infection in C57BL/6 and BALB/c mice. Infect. Immun. 68, 6946–6953 (2000).
Kindler, V., Sappino, A. P., Grau, G. E., Piguet, P. F. & Vassalli, P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 56, 731–740 (1989).
Flynn, J. L. et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572 (1995).
Jayaraman, P. et al. TIM3 mediates T cell exhaustion during mycobacterium tuberculosis infection. PLoS Pathog. 12, e1005490 (2016).
van Dissel, J. T. et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine 32, 7098–7107 (2014).
Famelis, N. et al. Architecture of the mycobacterial type VII secretion system. Nature 576, 321–325 (2019).
Tufariello, J. M. et al. Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc. Natl Acad. Sci. USA 113, E348–357 (2016).
Knudsen, N. P. et al. Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proc. Natl Acad. Sci. USA 111, 1096–1101 (2014).
Billeskov, R. et al. Difference in TB10.4 T-cell epitope recognition following immunization with recombinant TB10.4, BCG or infection with Mycobacterium tuberculosis. Eur. J. Immunol. 40, 1342–1354 (2010).
Rustad, T. R., Harrell, M. I., Liao, R. & Sherman, D. R. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 3, e1502 (2008).
Yu, G. et al. Gene expression analysis of two extensively drug-resistant tuberculosis isolates show that two-component response systems enhance drug resistance. Tuberculosis 95, 303–314 (2015).
Gideon, H. P. et al. Bioinformatic and empirical analysis of novel hypoxia-inducible targets of the human antituberculosis T cell response. J. Immunol. 189, 5867–5876 (2012).
Glickman, M. S., Cox, J. S. & Jacobs, W. R. Jr. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell 5, 717–727 (2000).
Hunter, R. L., Hwang, S. A., Jagannath, C. & Actor, J. K. Cord factor as an invisibility cloak? A hypothesis for asymptomatic TB persistence. Tuberculosis 101s, S2–s8 (2016).
Corrales, R. M. et al. Phosphorylation of mycobacterial PcaA inhibits mycolic acid cyclopropanation: consequences for intracellular survival and for phagosome maturation block. J. Biol. Chem. 287, 26187–26199 (2012).
Deng, G. et al. Identification of secreted O-mannosylated proteins from BCG and characterization of immunodominant antigens BCG_0470 and BCG_0980. Front. Microbiol. 11, 407 (2020).
Tima, H. G. et al. Inflammatory properties and adjuvant potential of synthetic glycolipids homologous to mycolate esters of the cell wall of Mycobacterium tuberculosis. J. Innate Immun. 9, 162–180 (2017).
Tanghe, A. et al. Improved immunogenicity and protective efficacy of a tuberculosis DNA vaccine encoding Ag85 by protein boosting. Infect. Immun. 69, 3041–3047 (2001).
Cocchi, M., Biancolillo, A. & Marini, F. Comprehensive Analytical Chemistry. Vol. 82 (eds Joaquim Jaumot, Carmen Bedia, & Romà Tauler) 265–299 (Elsevier, 2018).
Acknowledgements
We acknowledge EC HORIZON2020 TBVAC2020 (grant agreement no. 643381); EC ITN FP7 VACTRAIN project (the text represents the authors’ views and does not necessarily represent a position of the Commission who will not be liable for the use made of such information), The Netherlands Organization for Scientific Research (NWO-TOP Grant Agreement No. 91214038 and NWO-CHEM-TOPPUNT project grant agreement no. 21335).
Author information
Authors and Affiliations
Contributions
M.C., A.G., M.R. and T.H.M.O designed the study; M.C. performed the data analysis; T.H.M.O. gave insights on the analysis; K.L.M.C.F. performed the production and the purification of recombinant proteins; F.J. performed the experiments shown in Fig. 1 and in the Supplementary Fig. 1; H.G.T. contributed to experiments shown in Fig. 1; S.J.F. van den E. performed the experiment shown in Fig. 2; A.G. supervised the experiment shown in Fig. 2; M.R. supervised the experiments shown in Fig. 1 and in the Supplementary Fig. 1; M.C. and T.H.M.O. wrote the original draft and subsequent revised versions; A.G. and M.R. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Coppola, M., Jurion, F., van den Eeden, S.J.F. et al. In-vivo expressed Mycobacterium tuberculosis antigens recognised in three mouse strains after infection and BCG vaccination. npj Vaccines 6, 81 (2021). https://doi.org/10.1038/s41541-021-00343-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-021-00343-2
This article is cited by
-
Identification of differentially recognized T cell epitopes in the spectrum of tuberculosis infection
Nature Communications (2024)
-
Antigen identification strategies and preclinical evaluation models for advancing tuberculosis vaccine development
npj Vaccines (2024)
-
BCGΔBCG1419c increased memory CD8+ T cell-associated immunogenicity and mitigated pulmonary inflammation compared with BCG in a model of chronic tuberculosis
Scientific Reports (2022)
