
Abstract
Regulatory T cells (Treg) express the forkhead box p3 (Foxp3) transcription factor and suppress pathological immune responses against self and foreign antigens, including commensal microorganisms. Foxp3 has been proposed as a master key regulator for Treg, required for their differentiation, maintenance, and suppressive functions. Two types of Treg have been defined. Natural Treg (nTreg) are usually considered to be a separate sublineage arising during thymus differentiation. Induced Treg (iTreg) originate upon T cell receptor (TCR) stimulation in the presence of tumor growth factor β. Although under homeostatic conditions most Treg in the periphery are nTreg, special immune challenges in the intestine promote more frequently the generation of iTreg. Furthermore, recent observations have challenged the notion that Treg are a stable sublineage, and they suggest that, particularly under lymphopenic and/or inflammatory conditions, Treg may lose Foxp3 and/or acquire diverse effector functions, especially in the intestine, which may contribute to uncontrolled inflammation.
Similar content being viewed by others

Introduction
Progress in understanding the cell biology of regulatory T cells (Treg) came with the discovery of the X chromosome-encoded gene forkhead box p3 (Foxp3) during efforts to identify the genetic basis for the autoimmune disorder in human patients suffering from immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome and in the spontaneous mouse mutant scurfy.1, 2, 3 Mice harboring a loss-of-function mutation in the Foxp3 gene are affected by fatal early onset of lymphoproliferative immune-mediated disease involving multiple organs and tissues. Moreover, subsequent studies showed that sustained Foxp3 expression in mature Treg is required for their maintenance and suppressive function. Interestingly, diminished Foxp3 expression by Treg, as a result of gene targeting, led to the acquisition of effector T cell functions, including production of interleukin (IL)-2, IL-4, IL-17 and interferon (IFN)γ,4, 5 whereas forced expression of the Foxp3 gene in CD4+ non-Treg cells is sufficient to acquire suppressive function in vitro and in vivo.6, 7, 8 Together, these studies are consistent with the notion that Foxp3 has a pivotal role in defining Treg function.
Foxp3 induction in natural Treg (nTreg) occurs in vivo during thymic differentiation, under the influence of relatively high avidity interactions of the TCR with self-antigens9. Foxp3 can also be induced post-thymically, in conventional mature T lymphocytes when they are activated in vitro or in vivo in the presence of the cytokine tumor growth factor (TGF)-β.10, 11, 12, 13, 14 Nucleic acid sequence studies of the TCR α repertoire, in TCR β only transgenic mice, or secondary α rearrangements in TCR αβ transgenic mice, indicate that the TCR repertoire of peripheral Treg is more similar to thymic Treg than to conventional peripheral T cells. These data suggest, therefore, that under steady-state conditions most Foxp3-expressing lymphocytes in the spleen and blood are nTreg.15, 16 However, as discussed below, induced Treg (iTreg) are readily induced at the mucosal interface of the intestine. At present, it is neither certain how to distinguish nTreg from iTreg, nor is it established whether nTreg and iTreg can in some circumstances have different biological functions.
Although evidence indicates that thymic imprinted Treg are a stable sublineage,17, 18, 19, 20 in this review, we will discuss recent findings that suggest that populations of Treg, presumed to contain mostly, if not exclusively, nTreg can be plastic, meaning that they are capable of losing Foxp3 and their regulatory function, and, conversely, capable of acquiring various effector functions. Interestingly, the acquisition of specific effector functions may be more prevalent in T lymphocytes in the intestine.
Increased iTreg Formation in the Intestine
Several studies indicate that the majority of Treg present in the periphery are of thymic origin,15, 16 whereas, in contrast, there is evidence indicating that the small and large intestine and gut-associated lymphoid tissues, including the mesenteric lymph node (MLN), contain cells that preferentially promote iTreg differentiation. Consistent with this, TGF-β, required in most cases for the induction of Foxp3 by peripheral mature CD4+ T cells,10, 11, 12, 13 is highly enriched in the intestine. In vivo studies show that the gut-associated lymphoid tissue is a pivotal anatomical site for the induction of Foxp3 expression under conditions of chronic antigen exposure, and that this is crucial for the maintenance of tolerogenic conditions.11, 21, 22, 23 Agents that drive iTreg generation include food antigens, which are modeled in oral tolerance experiments, and recently the diet-derived vitamin A metabolite, retinoic acid (RA), was identified as a critical regulator of TGF-β-dependent differentiation. Whereas RA can have direct cell intrinsic effects in naïve CD4 T cells by suppressing RORγt and IL-17, and conversely by enhancing Foxp3 induction,24, 25, 26, 27 RA might further also indirectly augment iTreg formation by acting on effector T cells, through inhibition of production of cytokines, such as IL-4 and IFNγ, that prevent Foxp3 expression.28 Among dendritic cells, mucosal CD103+ dendritic cells have been reported to release RA, leading to enhanced induction of Foxp3 by the naïve CD4+ T cells they prime.24, 29, 30, 31, 32 Formation of Treg in vitro under these conditions led to a Treg population that was particularly effective at preventing disease in the T-cell transfer model of colitis.24 Moreover, macrophages in the small intestine lamina propria are also able to convert naïve T lymphocytes into Foxp3+ Treg in an IL-10-, RA- and TGF-β-dependent manner.33
Besides, the results from several studies of germ-free mice indicate that the absence or presence of microbial antigens from commensal organisms modulate the number and function of Treg in the intestine. Although the components of the flora responsible for this have not been fully defined,34, 35, 36 studies show that a segmented filamentous bacterium can influence the number of Treg.37, 38 It remains to be determined, however, if the microflora acts mainly on iTreg generation, nTreg homeostasis, or on both processes.
Furthermore, the number of Foxp3 expressing Treg in the small intestine lamina propria is increased in mice deficient for TLR9, which senses nucleic acids containing unmethylated CpG dinucleotides that are characteristic of microbes.23 The formation of iTreg in co-cultures of naïve T cells with lamina propria dendritic cells was suppressed by DNA from the gut flora, suggesting that the increased formation of Treg in the lamina propria in Tlr9−/− mice is due to increased formation of iTreg.23
Epigenetic Modifications of Foxp3 in nTreg
Studies have shown that nTreg are relatively stable in a healthy immune system, and that Foxp3 has an indispensable role in maintaining the intrinsic stability of Treg. Foxp3 maintains a positive auto-feedback loop, and the disruption of Foxp3 protein function in committed Treg resulted in reduced foxp3 transcription.5 Ablation of Cbfβ, a cofactor of the Runt-related transcription factor 1, which binds to the Foxp3 protein, led to decreased expression of Foxp3 in Treg and impaired suppressive function in vitro and in vivo.39 Moreover, knockdown of Runt-related transcription factor 1 showed that Runt-related transcription factor 1 is required for the optimal regulation of Foxp3 expression in human T cells.39
Several reports suggest that epigenetic modifications are also key factors for the stability of Foxp3 expression.17, 18, 19, 20, 40, 41, 42 Stable Foxp3 expression by Treg is associated with selective demethylation of evolutionarily conserved CpG-rich elements within the Foxp3 locus.17, 18, 19, 20 Consistent with Foxp3 auto-regulation, it was recently shown that Foxp3 can bind to a conserved non-coding DNA sequence (CNS) element 2 (CNS2), and that CNS2 controls the heritable maintenance of Foxp3 expression in a manner that is dependent on Cbfβ-Runt-related transcription factor 1 activity and CpG DNA demethylation.43 This suggests that Foxp3 recruitment to this “cellular memory module” facilitates the heritable maintenance of the active state of the Foxp3 locus and, subsequently, Treg lineage stability.43 In contrast, other Foxp3 CNS elements have different roles. CNS1 controls peripheral, but not thymic, induction of Foxp3 expression, while CNS3 controls de novo Foxp3 expression in the thymus. Interestingly, in vitro TGFβ-induced Foxp3+ T cells retain a different pattern of demethylated CpG motifs in the CNS2 region, which could be related to the stability difference between the more stable nTreg and iTreg.17, 18, 19, 44 Taken together, there is a strong correlation between stable Foxp3 expressions, the consequent function of Treg, and a more complete demethylated foxp3 gene status. However, it is still not known what molecules initiate and regulate these modifications, and how Foxp3 itself is involved in this process.
Loss of Foxp3 Expression by Treg
Although there is no surface marker that can distinguish nTreg from iTreg with certainty, as noted above, there is evidence that Foxp3 expressing cells from spleen, lymph nodes, and other sites, not including the intestine, are mostly nTreg. Although epigenetic modifications indicate mechanisms for maintenance of stable expression of the foxp3 gene, other data suggest the plasticity of Treg populations, including predominantly nTreg. There are several reports that these presumed nTreg lose Foxp3 expression in vitro, for example when the TCR of the cells is engaged in the presence of IL-6.41, 45 Furthermore, an agonist antibody to the T-cell Ig mucin 1 caused loss of Foxp3 expression and loss of effector function.46 In addition, several recent reports indicate that nitric oxide can reduce Foxp3 expression by splenic T cells, but it remains uncertain whether this in fact causes the loss of Foxp3 expression by Treg, as opposed to decreased homeostasis or survival, and the in vivo consequences on Treg function remain to be explored.47, 48
In vivo data for the instability of Treg originate predominantly from studies involving T-cell transfer to lymphopenic mice. Most Treg retain high Foxp3 expression following adoptive transfer to immune competent mice.17, 49 However, up to 50% of Treg may lose Foxp3 expression after adoptive transfer into lymphopenic hosts.49, 50 In transfers of Treg alone, the loss of Foxp3 was attributed to reduced IL-2 production in the absence of accompanying effector cells. It still remains possible in these transfers that following multiple rounds of proliferation a minor contaminant undergoes selective expansion, thereby accounting for the phenotypic changes in transferred Foxp3+ cells. Perhaps more likely however, there could be some heterogeneity in the Foxp3+ Treg population. In fact, a recent study showing 50% loss of Foxp3 attributed this to the presence of a CD25low subset of Foxp3+ cells that is less stable but more capable of expanding under lymphopenic conditions in these Rag2−/− mice.51 Upon losing Foxp3, the “lapsed Treg” in this study no longer expressed high levels of other Treg markers such as GITR and CTLA-4, and they were unable to suppress T effector cell proliferation in vitro.
In the CD4+CD45RBhigh T-cell transfer model of colitis, it is well known that Foxp3+ cells can not only prevent, but also cure disease.52, 53, 54 These Treg were not effective, however, in the absence of IL-10 signals, and Treg deficient for the expression of the IL-10 receptor β chain or Treg transferred to lymphopenic IL-10 deficient Rag−/− hosts lost Foxp3 expression and failed to control disease.55 In this case, sorting for CD25 high cells, as well as for Foxp3 promoter driven gene reporter expression, ruled out the selective outgrowth of a CD25low Treg subset. Further experiments also discard the potential selective outgrowth of either effector or memory CD4+ T cells, providing evidence for a true loss of Foxp3 expression by Treg. This work established a role for paracrine Il-10 from intestinal myeloid cells in the maintenance of Treg function. By analyzing the cells from Rag−/− mice that also have an Il-10 gene reporter construct, it was found that myeloid cells in the intestine lamina propria, but not in the spleen or MLN, have constitutive expression of IL-10. At 1 week after the injection of CD4+ T cells, however, the number of reporter gene positive cells decreased in the large intestine lamina propria but increased in the MLN, suggesting a migration of this population from the lamina propria to the MLN in the presence of activated T lymphocytes. These results were confirmed by the analysis of Il-10 mRNA in Rag−/− mice without the reporter construct. As noted above,33 myeloid cells that constitutively produce IL-10 have also been described in the small intestine lamina propria, and were reported to be important for iTreg generation.
On analyzing the gene reporter mice, IL-10 producing myeloid cells were found only in the lamina propria before T-cell injection, and in the MLN afterwards. Despite this, the loss of Foxp3 expression by the transferred Treg in the absence of IL-10 was systemic, although it was slightly more pronounced in the intestine. The systemic loss of Foxp3 implies, however, that the transferred Treg found in the spleen and elsewhere at some point had contact with IL-10 producing myeloid cells. Alternatively, the IL-10 from the intestinal cells could act systemically. A model we propose for these results is that colitis preventing Treg are primed mostly in the MLN, and under the influence of IL-10 producing antigen presenting cells that help to maintain Foxp3 expression. The Treg will eventually lose Foxp3, however, if IL-10 is not present during the crucial priming events. Regardless of the presence of IL-10, the primed Treg recirculate systemically in the lymphopenic recipients. This model can account for the systemic loss of Foxp3 expression due to the IL-10 synthesis defect in intestinal antigen presenting cells.
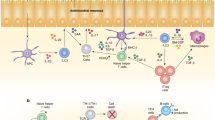
Consistent with the pivotal role of myeloid cells from the lamina propria, transfer of a CD11b+CD11c+F4/80+ population from IL-10 sufficient mice prevented the loss of Foxp3 expression by Treg in the spleen of Il10−/−Rag−/− recipients. Interestingly, the loss of Foxp3 expression occurred only in recipients with colitis, indicating that the requirement for IL-10 is manifested only in the presence of inflammation (Figure 1).

Conditions leading to maintenance and loss of regulatory T cells (Treg) in the intestine and elsewhere are illustrated. Top: Interleukin (IL)-2 maintains Treg populations systemically, and Treg may be either induced or under inflammatory conditions maintained by IL-10 from macrophages or other myeloid cells in intestinal tissues. Bottom: IL-12 may lead to induction of T-bet expression in the intestine, and it, or other inflammatory cytokines, may contribute to the loss of Foxp3 expression when Treg are transferred to lymphopenic mice that lack IL-10.
Consistent with the plasticity of Treg cells, in Cd3ɛ−/− mice, which still contain B lymphocytes, transferred Treg gave rise to follicular B helper T cells (TFH) under the influence of CD40 expressed by B cells.56 The conversion of Foxp3 Treg to TFH cells apparently occurred only in the Peyer's patches, demonstrating an intestinal mucosa-specific adaptation of the Treg population.
Instability of Treg cells has also been observed in an immune competent environment. Following infection with Toxoplasma gondii, for example, the number of Treg dramatically decreased due to impaired induction, as well as loss of existing Treg.57 These data suggest impaired homeostasis of the Treg, rather than plasticity or true loss of function. However, particularly in the small intestine lamina propria, cells that retained Foxp3 also expressed T-bet and acquired the ability to produce IFNγ.57
Results obtained from a fate mapping system, designed to detect cells that previously expressed Foxp3, also provide evidence for the plasticity of Treg, although this was not specific to the intestine. This system used mice transgenic for a Foxp3–GFP–Cre fusion protein crossed to a ROSA26-YFP mouse with a floxed stop transcription signal. Cre activity in these mice causes the expression of YFP, and lapsed Treg, or those that have lost Foxp3 expression, can be identified as YFP+GFP− cells. Whereas relatively few cells lost Foxp3 expression under homeostatic conditions in the Peyer's patches and elsewhere, when crossed onto the autoimmune nonobese diabetic background, a significantly increased proportion of the cells became Foxp3 negative.58
Lapsed Treg Acquire Diverse Effector Functions
Interestingly, the lapsed Treg acquire effector functions. For example, Treg activated in vitro in the presence of IL-6 lost Foxp3 expression and differentiated into IL-17 producing cells.45, 59 The effector functions these cells may acquire in vivo are diverse, as summarized in Table 1. In one study, adoptive transfer of Treg alone into lymphopenic hosts resulted in the loss of Foxp3 expression and those lapsed Treg acquired the ability to produce several proinflammatory cytokines, including IFNγ and IL-17.48, 49 In other studies, when lymphopenic recipients were immunized with myelin oligodendrocyte glycoprotein peptide and CFA, a significant percentage of Treg lost Foxp3 expression and the lapsed Treg converted preferentially to IL-17 producing cells.59, 60 Interestingly, Foxp3 and IL-17 double expressers were also observed in this situation.59 We found that in lymphopenic hosts that lack IL-10 production those transferred Treg that lost Foxp3 and suppressive function also developed into IFNγ producing “Th1 effector like” cells that expressed T-bet.55 This change in the Treg population was driven by the inflammatory environment in the recipient mice, and therefore the strong Th1 like phenotype of the lapsed cells may reflect the action of one or more inflammatory cytokines. The Th1 like phenotype of the lapsed Treg bears some resemblance to the phenotype of altered intestinal Treg following T. gondii infection, but these cells tended to maintain Foxp3 expression along with T-bet and the ability to produce IFNγ. By contrast, in the absence of colitis, the adoptive transfer of Treg into Cd3ɛ−/− hosts, which retain B lymphocytes, resulted in the loss of Foxp3 expression and the generation of lapsed Treg that differentiated into TFH cells in Peyer's patches that promoted IgA class switching.56
Furthermore, lapsed Treg that arose in immune competent mice, especially when crossed to the autoimmune susceptible nonobese diabetic strain genetic background, also acquired an effector memory phenotype, with heterogeneous CD62L expression and high CD44 expression. Furthermore, these cells produced several proinflammatory cytokines, such as IFNγ and IL-17, rather than a focused Th1 like cytokine pattern. Adoptive transfer of the lapsed Treg led to the rapid onset of diabetes, indicating that these cells could trigger autoimmunity.58
Collectively, the results indicate that when Treg lose Foxp3, which can occur under lymphopenic and/or certain inflammatory conditions, they do not simply become anergic T cells but instead develop into effector T cells that might have pivotal roles in inflammation and autoimmunity. Furthermore, in the intestine these cells may have unique roles, such as differentiation into TFH cells.
Mechanisms for the Acquisition of New Functions
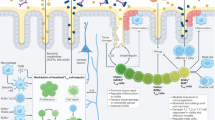
The studies summarized above suggest that Foxp3 cells are able to acquire effector functions when the restraint of Foxp3 is removed. Consistent with this hypothesis, it was shown that Foxp3+ cells from spleen and lymph node, containing predominantly nTreg, lack a suppressive chromatin marker, histone 3 lysine 27 trimethylation, in the Ifnγ locus. Other CD4+ T-cell subsets, however, including even iTreg generated in vitro, show some histone 3 lysine 27 trimethylation, suggesting that IFNγ is less repressed in nTreg. In addition, the gene encoding the signature Th1 transcription factor T-bet exhibited a broad spectrum of epigenetic states in the Foxp3+ cells from spleen and lymph node, consistent with the ability to induce T-bet and IFNγ expression in vitro in these Treg populations under Th1 conditions.41
How do apparently lapsed Treg gain different kinds of effector functions? In this regard, it is interesting that several reports suggest that there are multiple, functional Treg subsets. One of these expresses T-bet, and it is specifically adapted for suppressing Th1 responses.61 Moreover, after T. gondii infection, the induction of expression of T-bet and IFNγ in Treg that retained Foxp3 expression was dependent in part on Th1 conditions. IFNγ was important for inducing T-bet expression by Treg, whereas IL-12 was most critical for inducing IFNγ synthesis by these cells.57 Another Treg subset was described to express a transcription factor essential for Th2 differentiation, interferon regulatory factor 4, which is a Foxp3 target gene.62 The absence of interferon regulatory factor 4 expressing Foxp3+ Treg, achieved by conditional ablation of the interferon regulatory factor 4 gene in these cells, resulted in a spontaneous induction of Th2 mediated inflammation.62 A third Treg subset is most important for repressing Th17 responses.63 It was found that STAT3 (signal transducer and activator of transcription 3), required for Th17 cells, is also required in Treg for the suppression of Th17 responses. In fact, mice with a Treg specific deficiency in STAT3 have increased Th17 cells and they develop a fatal colitis.63 In Treg, not only does STAT3 promote the expression of certain suppressive molecules, but it also interacts with Foxp3 to suppress the expression of molecules that promote Th17 development, for example the IL-6 receptor, which on wild-type Treg might allow these cells to compete for IL-6.63 Therefore, while Treg might respond to environmental cues, causing subsets to acquire transcriptional programs that allow them to localize with and regulate subsets of effectors, in the context of the loss of Foxp3 expression, they might instead synergize with and resemble these effector subsets.
Conclusions
In summary, a number of experiments indicate a degree of plasticity in Foxp3+ Treg and these cells can acquire diverse effector functions, some of which are intestine specific, such as the TFH function found in Treg accumulating in the Peyer's patches. Interestingly, much of the evidence for Treg obtaining effector functions was observed in studies in which colitis was induced following cell transfer to lymphopenic mice. For reasons that are not known, in these experiments the loss of Foxp3 expression tended to be more pronounced in intestinal tissues. Therefore, some factor(s) known to be important for Treg homeostasis, such as IL-2 production or CD28 mediated costimulation,64 may be limiting in the intestine or, alternatively, gut-specific cues might promote the conversion of Treg cells to effector cells under certain conditions. It seems counterintuitive, however, that self-reactive Treg, or Treg specific for dietary antigens or antigens from the commensal flora, would easily lose regulatory function and become effector cells, as this would present a clear danger to the organism. It should be emphasized that this loss of Foxp3 expression was most readily observed following cell transfer to lymphopenic mice, and/or under inflammatory conditions. In fact, in our own transfer experiments to lymphopenic mice, the substantial loss of Foxp3 expression and regulatory function required the coordinate action of two factors, namely, the loss of IL-10 signaling to the Treg and the presence of an inflammatory state in the host. Therefore, while plasticity in this population can be detected, we would agree that the maintenance of the Treg phenotype is favored under immune quiescent conditions. The types of physiological conditions that might induce Treg plasticity remain to be fully defined.
Another issue is whether the Foxp3+ cells exhibiting the greatest plasticity are a subset that may expand preferentially under some conditions. One example of such a subset are the Foxp3+ cells that have low expression of CD25, which preferentially expanded under lymphopenic conditions and tended to lose Foxp3 expression.51 In our experiments in which IL-10 was deprived, however, the CD25low subset of Foxp3+ cells was excluded. We cannot rule out the possibility, however, that there is heterogeneity even among the CD25high Foxp3+ cells, and that a subset of these cells might expand preferentially in lymphopenic mice with colitis, in the absence of IL-10. For example, a minority of iTreg in the CD25high Foxp3+ population from spleen and lymph node might expand preferentially following transfer. The assumption underlying this hypothesis, however, is that such hyper expanding iTreg are also less stable. While epigenetic modifications of the Foxp3 locus in iTreg are consistent with a reduced stability, due to increased methylation,64 it remains to be more rigorously analyzed whether iTreg, or populations presumed to be enriched for iTreg from the intestine, are in fact less stable after transfer.
It is now critical to identify markers for those Foxp3+ cells that are most likely to lose Foxp3 expression, and for those cells that have already lost Foxp3. If this could be done, one would hope to extend these findings to human Foxp3+ Treg, although it must be acknowledged that there are differences in Foxp3 expression by T cells on comparing humans and mice.65 Nevertheless, a major goal will be to determine whether the plasticity of Treg has a role in inflammatory bowel disease and other inflammatory and immune pathologies in humans. In addition, it will be particularly important, from both the theoretical and therapeutic viewpoints, to determine whether the lapsed cells retain significant portions of the Treg transcriptional signature, and whether they can be rehabilitated to regain Treg function.
References
Bennett, C.L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21 (2001).
Brunkow, M.E. et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73 (2001).
Wildin, R.S. et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20 (2001).
Wan, Y.Y. & Flavell, R.A. Regulatory T cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445, 766–770 (2007).
Williams, L.M. & Rudensky, A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 8, 277–284 (2007).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science (New York, NY) 299, 1057–1061 (2003).
Fontenot, J.D., Gavin, M.A. & Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336 (2003).
Khattri, R., Cox, T., Yasayko, S.A. & Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342 (2003).
Picca, C.C., Larkin, J. III, Boesteanu, A., Lerman, M.A., Rankin, A.L. & Caton, A.J. Role of TCR specificity in CD4+ CD25+ regulatory T cell selection. Immunol. Rev. 212, 74–85 (2006).
Chen, W. et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 (2003).
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M.C. & von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227 (2005).
Selvaraj, R.K. & Geiger, T.L. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J. Immunol. 179 11 p following 1390 (2007).
Zheng, S.G., Wang, J.H., Gray, J.D., Soucier, H. & Horwitz, D.A. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J. Immunol. 172, 5213–5221 (2004).
Mucida, D., Kutchukhidze, N., Erazo, A., Russo, M., Lafaille, J.J. & Curotto de Lafaille, M.A. Oral tolerance in the absence of naturally occurring Tregs. J. Clin. Invest. 115, 1923–1933 (2005).
Hsieh, C.S., Zheng, Y., Liang, Y., Fontenot, J.D. & Rudensky, A.Y. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat. Immunol. 7, 401–410 (2006).
Wong, J., Mathis, D. & Benoist, C. TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J. Exp. Med. 204, 2039–2045 (2007).
Floess, S. et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5, e38 (2007).
Polansky, J.K. et al. DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 38, 1654–1663 (2008).
Lal, G. et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 182, 259–273 (2009).
Lal, G. & Bromberg, J.S. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 114, 3727–3735 (2009).
Curotto de Lafaille, M.A., Kutchukhidze, N., Shen, S., Ding, Y., Yee, H. & Lafaille, J.J. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126 (2008).
Belkaid, Y. & Oldenhove, G. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity 29, 362–371 (2008).
Hall, J.A. et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 29, 637–649 (2008).
Mucida, D. et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (New York, NY) 317, 256–260 (2007).
Elias, K.M. et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111, 1013–1020 (2008).
Xiao, S. et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol. 181, 2277–2284 (2008).
Mucida, D. et al. Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity 30, 471–472; author reply 472–473 (2009).
Hill, J.A. et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity 29, 758–770 (2008).
Annacker, O. et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061 (2005).
Benson, M.J., Pino-Lagos, K., Rosemblatt, M. & Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 204, 1765–1774 (2007).
Coombes, J.L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Sun, C.M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 Treg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Denning, T.L., Wang, Y.C., Patel, S.R., Williams, I.R. & Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086–1094 (2007).
Strauch, U.G. et al. Influence of intestinal bacteria on induction of regulatory T cells: lessons from a transfer model of colitis. Gut 54, 1546–1552 (2005).
Ostman, S., Rask, C., Wold, A.E., Hultkrantz, S. & Telemo, E. Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 36, 2336–2346 (2006).
Ishikawa, H. et al. Effect of intestinal microbiota on the induction of regulatory CD25+ CD4+ T cells. Clin. Exp. Immunol. 153, 127–135 (2008).
Gaboriau-Routhiau, V. et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31, 677–689 (2009).
Ivanov, II. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Kitoh, A. et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity 31, 609–620 (2009).
Kim, H.P. & Leonard, W.J. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J. Exp. Med. 204, 1543–1551 (2007).
Wei, G. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009).
Huehn, J., Polansky, J.K. & Hamann, A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T cell lineage? Nat. Rev. Immunol. 9, 83–89 (2009).
Zheng, Y., Josefowicz, S., Chaudhry, A., Peng, X.P., Forbush, K. & Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T cell fate. Nature 463, 808–812 (2010).
Baron, U. et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur. J. Immunol. 37, 2378–2389 (2007).
Xu, L., Kitani, A., Fuss, I. & Strober, W. Cutting edge: regulatory T cells induce CD4+CD25−Foxp3− T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 178, 6725–6729 (2007).
Degauque, N. et al. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J. Clin. Invest. 118, 735–741 (2008).
Brahmachari, S. & Pahan, K. Myelin basic protein priming reduces the expression of Foxp3 in T cells via nitric oxide. J. Immunol. 184, 1799–1809 (2010).
Brahmachari, S. & Pahan, K. Suppression of regulatory T cells by IL-12p40 homodimer via nitric oxide. J. Immunol. 183, 2045–2058 (2009).
Gavin, M.A. et al. Foxp3-dependent programme of regulatory T cell differentiation. Nature 445, 771–775 (2007).
Duarte, J.H., Zelenay, S., Bergman, M.L., Martins, A.C. & Demengeot, J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur. J. Immunol. 39, 948–955 (2009).
Komatsu, N., Mariotti-Ferrandiz, M.E., Wang, Y., Malissen, B., Waldmann, H. & Hori, S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA 106, 1903–1908 (2009).
Powrie, F., Correa-Oliveira, R., Mauze, S. & Coffman, R.L. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J. Exp. Med. 179, 589–600 (1994).
Mottet, C., Uhlig, H.H. & Powrie, F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 (2003).
Uhlig, H.H. et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 177, 5852–5860 (2006).
Murai, M. et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178–1184 (2009).
Tsuji, M. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science (New York, NY) 323, 1488–1492 (2009).
Oldenhove, G. et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786 (2009).
Zhou, X. et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007 (2009).
Yang, X.O. et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56 (2008).
Korn, T. et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 13, 423–431 (2007).
Koch, M.A., Tucker-Heard, G., Perdue, N.R., Killebrew, J.R., Urdahl, K.B. & Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602 (2009).
Zheng, Y. et al. Regulatory T cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356 (2009).
Chaudhry, A. et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (New York, NY) 326, 986–991 (2009).
Josefowicz, S.Z. & Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625 (2009).
Ziegler, S.F. FOXP3: of mice and men. Annu. Rev. Immunol. 24, 209–226 (2006).
Acknowledgements
This work was supported by the National Institutes of Health Grant PO1 DK46763 and a Senior Research Award from the Crohn's & Colitis Foundation of America to MK, National Institutes of Health Grants RO1 AI50265 to HC, a Career Development Award from the Crohn's & Colitis Foundation of America to MM, and a Deutsche Forschungsgemeinschaft fellowship to PK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Murai, M., Krause, P., Cheroutre, H. et al. Regulatory T-cell stability and plasticity in mucosal and systemic immune systems. Mucosal Immunol 3, 443–449 (2010). https://doi.org/10.1038/mi.2010.27
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.27
This article is cited by
-
The immunomodulatory effects of long-term supplementation with Lactobacillus casei Shirota depend on ovalbumin presentation in BALB/c mice
Scientific Reports (2021)
-
CD69 enhances immunosuppressive function of regulatory T-cells and attenuates colitis by prompting IL-10 production
Cell Death & Disease (2018)
-
Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients
Scientific Reports (2016)
-
Development and maintenance of intestinal regulatory T cells
Nature Reviews Immunology (2016)
-
Epigenetic control of Foxp3 by SMYD3 H3K4 histone methyltransferase controls iTreg development and regulates pathogenic T-cell responses during pulmonary viral infection
Mucosal Immunology (2015)
