
Abstract
The production of immunoglobulin A (IgA) in mammals exceeds all other isotypes, and it is mostly exported across mucous membranes. The discovery of IgA and the realization that it dominates humoral mucosal immunity, in contrast to the IgG dominance of the systemic immune system, was early evidence for the distinct nature of mucosal immunology. It is now clear that IgA can function in high-affinity modes for neutralization of toxins and pathogenic microbes, and as a low-affinity system to contain the dense commensal microbiota within the intestinal lumen. The basic map of induction of IgA B cells in the Peyer's patches, which then circulate through the lymph and bloodstream to seed the mucosa with precursors of plasma cells that produce dimeric IgA for export through the intestinal epithelium, has been known for more than 30 years. In this review, we discuss the mechanisms underlying selective IgA induction of mucosal B cells for IgA production and the immune geography of their homing characteristics. We also review the functionality of secretory IgA directed against both commensal organisms and pathogens.
Similar content being viewed by others


Introduction
Historical aspects of IgA and the mucosal immune system
It is very appropriate that there should be a review of immunoglobulin A (IgA) in this first issue of Mucosal Immunology. We can justify our discipline (and our journal) on the distinct features of immunity at mucosal surfaces, and classical studies of the IgA isotype yielded some of the first evidence for this.
The γA class of immunoglobulins was discovered by Gugler, Heremans, and co-workers from the observations that not all myeloma proteins were accounted for by the 7S (low carbohydrate content) and the 16S (high carbohydrate) classes. Subsequently, an antigenically distinct immunoglobulin with a high carbohydrate content named γA (or later IgA) that migrates in the β-globulin region after electrophoresis was characterized.1, 2 This isotype was subsequently shown to be predominant in exocrine secretions.3, 4 More than eight models were proposed in the late 1960s and early 1970s to explain how IgA selectively could reach the secretions. One of us suggested in 1974 that the epithelial glycoprotein identified by Tomasi et al.5 as part of secretory IgA (SIgA) and later on named secretory component (SC) by WHO could act as a membrane receptor for dimeric IgA and pentameric IgM on secretory epithelium.6 This model formed the basis for a common epithelial transport mechanism to generate SIgA and SIgM, in which SC and the joining (J) chain of polymeric Ig (pIgs) constituted “key and lock” molecules.7 This model is now generally accepted (see the section Selective epithelial export of locally produced IgA and IgM) and membrane SC has been termed pIg receptor (pIgR).
Studies that compared humoral immunity at mucosal surfaces with serum immune responses had repeatedly shown a separation between secretory and systemic immune responses (see Table 1; also reviewed in ref. 8). In general, immunization or infection at mucous membranes resulted in high titer of protective antibodies at the mucosal site with absent or low titers in serum, whereas the reverse occurred with parenteral immunization. In retrospect, not all the locally induced antibodies detected in these studies would have been of the IgA class, but the facts of (i) a distinct mucosal immune response at mucosal surfaces and (ii) a distinct Ig isotype that dominated mucosal antibodies constituted strong evidence for a local immune system structurally and functionally distinct from the systemic compartment.
We now know that although IgA is a relatively small component of serum antibodies, the abundance of IgA-secreting cells in normal mucosae means that this isotype actually comprises at least 70% of all Ig produced in mammals. Most T cells are also found at mucosal surfaces.
IgA induction by commensal intestinal microbes
This dominance of IgA production in the intestinal mucosa depends on colonization with environmental microbes. While microbes in soils, subsurfaces, and oceans of the earth generally reach a density of ⩽108 organisms per g, the mammalian lower intestine is an extraordinarily good habitat where bacteria achieve densities of up to 1012 organisms per g contents.9 One of the most dramatic and first discovered adaptations of the host to the presence of these microbes was the fact that mucosal IgA-secreting cells are drastically reduced in germ-free animals, and are virtually absent in neonates before they have been colonized with a commensal microflora.10, 11, 12 Although intestinal IgA is clearly induced by the presence of commensal intestinal microbes, the function of the IgA response in maintaining mutualism between host and microbe is far less clear. Particularly intriguing is the fact that selective IgA deficiency is a common immunodeficiency with a generally mild phenotype.13 These functional aspects are discussed further in the section Function of IgA in different systems of this review.
IgA neutralization of pathogens and exotoxins
The other important set of classical observations showed that IgA can act as a neutralizing antibody to pathogens and exotoxins. For example, cholera toxin has been very widely used as a model immunogen for in vivo IgA induction, and SIgA can protect from toxin-induced fluid accumulation in a ligated intestinal loop model.14 Usually the formal affinities for toxin and pathogen-protective effects are unknown, but comparisons with neutralizing antibodies of other classes suggest that these antibodies probably work at 10−9/M or better.15 In some cases, the neutralization effect occurs during transport of IgA through the epithelial cell layer,16, 17 and the mechanisms and affinities here may be different. The general principle is that IgA appears to function across a spectrum of near innate immune responses, as described in the section IgA induction by commensal intestinal microbes, and highly adaptive responses as evidenced by the functional neutralization of pathogenic molecules and microbes.
IgA Class Switch Recombination Within and Outside the Intestinal Mucosa
In most mammals, a single gene encodes the α heavy chain (CHα) domains at the 3′ end of the constant Ig heavy chain locus. In humans and higher primates, however, there has been a duplication of the γ → α region, resulting in two functional CHα gene loci, encoding the IgA1 and IgA2 subclasses. The predominant difference between these two isotypes lies in the hinge region. Human IgA2 is more resistant to proteases because of a 13-amino acid deletion in the hinge region that removes the recognition site for IgA1-specific proteases. IgA2-producing plasma cells are enriched at secretory effector sites and dominate in the distal gut,18 whereas IgA1 is the main IgA subclass produced by the systemic immune system. Lagomorphs, such as rabbits, have multiple duplications of the CHα gene, resulting in 13 IgA isotypes. Generally, mammalian α loci have three exons corresponding to the constant heavy chain domains, with the region coding the 18-amino-acid extension of the secreted form at the 3′ end of the third exon (called the secretory tailpiece) and another short exon coding the transmembrane segment and cytoplasmic tail (Figure 1a). The tailpieces of IgA and IgM heavy chains allow selective combination of these isotypes with the J chain (see the section Selective epithelial export of locally produced IgA and IgM).

IgA structure and class switch recombination (CSR). (a) The intron–exon structure of the constant α heavy-chain (CHα) gene. Each exon encodes a separate domain (Cα1, Cα2, Cα3). The hinge (H) region is encoded at the 5′ end of the Cα2 exon and the 18-amino-acid extension at the C-terminal end of secreted IgA is encoded at the 3′ end of the Cα3 exon (tail piece available for J-chain combination). The transmembrane and cytoplasmic tail sequences are encoded by the αM exon located 3′ of the Cα3 exon. The shaded box represents 3′-untranslated RNA. (b) Three-dimensional model of monomeric human IgA2 showing wide separation of antigen-binding sites at the top (reproduced with permission from ref. 128). (c) Following V(D)J recombination, synthesis of the μ heavy chain and subsequent association with a light chain, an IgM molecule is expressed on the surface of the B cell. CSR to IgA (shown for the mouse) is preceded by the production of an α germline transcript (not shown), which is formed following transcription through the initiation (I) exon, the intronic switch (S) region, and the Cα exons. Transcription is initiated from cytokine-inducible promoters upstream of the I exon. Processing of this primary transcript generates the germline transcript and a spliced out S-region transcript. The spliced out S-region stably associates with the template DNA strand of the S-region, forming a stable RNA–DNA hybrid. The displaced G-rich single-stranded DNA is thought to be the substrate for the enzyme activation-induced cytidine deaminase (AID). DNA double-strand breaks at the S-region and DNA repair result in joining of the Sα and Sμ regions yielding the rearranged IgA heavy chain constant region and the excised intervening DNA, depicted as a DNA switch circle, which represents a short-lasting sign of local CSR. Eμ upstream of the Sμ region indicates the intronic enhancer. Gene elements depicted are not to scale.
A model of the three-dimensional structure of monomeric IgA2 is depicted in Figure 1b, showing the wide separation of the two antigen-binding fragments at the top. This unique characteristic of IgA probably provides this isotype with better binding properties in the face of large antigens such as bacteria.
Class switch recombination (CSR) to IgA occurs following transcription from the intronic α (Iα) promoter 5′ of the α locus,19 and requires the activation-induced cytidine deaminase20 (Figure 1c). The circular spliced segment is usually from the region between the switch segments upstream of the μ and α loci, although trans splicing between different chromosomes is also possible.21
In general, CSR has been found to require two signals. The first is through cytokines, which activate transcription of the promoters upstream of the CH exons and the switch region preceding them. The second is delivered by ligation of CD40 on B cells with its ligand CD40L on T cells. However, in mice and humans deficient in CD40L or CD40, IgA production is not abrogated.22, 23 The major cytokine signal for α-CSR is transforming growth factor (TGF)β with contributions from interleukin (IL)-2, IL-4, IL-5, IL-6, and IL-10, as initially demonstrated by studying IgA production in nonspecifically stimulated B-cell cultures24 (reviewed in ref. 25). Evidence for the role of TGFβ in vivo comes from an almost complete absence of IgA in mice deficient for the TGFβ receptor TβRII.26 Mice deficient for the TGFβ SMAD7 negative regulator have increased CSR to IgA but reduced proliferation to LPS,27 whereas mice deficient for the positive signal SMAD2 have no IgA.28 IL-4-deficient mice have normal total IgA levels, but an impaired specific response to mucosal immunization with cholera toxin,29 while IL-6-deficient mice have defective IgA responses in some systems.30, 31, 32
Because IgA production is partially independent of T cells33 and CD40-CD40L engagement, other costimulatory signals for α-CSR in B cells have been sought. Two members of the tumor necrosis factor (TNF) family—BAFF (B-cell activating factor of the TNF family, also known as BLyS (B-lymphocyte stimulator) in humans) and APRIL (A proliferation-inducing ligand)—have been shown to stimulate CSR to IgG and IgA in both human34 and mouse35 B cells. Monocytes and dendritic cells express BAFF, whereas APRIL is expressed by monocytes, macrophages, dendritic cells, and activated T cells.36 Recently, He et al.37 showed that in response to Toll-like receptor stimulation, human colonic epithelial cells also secrete BAFF and APRIL, which could lead to local switching to IgA within the human colon. B cells express all three receptors (BAFF-R, BCMA (B-cell maturation antigen), and TACI (transmembrane activator and CAML interactor)) for these cytokines (Figure 2). While APRIL binds only BCMA and TACI, BAFF can interact with all three receptors. However, APRIL, but not BAFF, can also bind to proteoglycans, such as syndecan, thereby providing an APRIL-specific binding partner.38

Costimulatory signals for class switch recombination (CSR) to IgA. BAFF and APRIL are expressed by dendritic cells, monocytes, and human colonic epithelial cells (following Toll-like receptor signaling, not shown), whereas APRIL can also be secreted by macrophages and activated T cells. These cytokines bind to their receptors, BAFF-R, TACI, or BCMA, which are all expressed by B cells. APRIL, A proliferation-inducing ligand; BAFF, B-cell activating factor of the TNF family; BCMA, B-cell maturation antigen; TACI, transmembrane activator and CAML interactor.
The function of these interactions has been investigated with mouse strains having selective deficiencies. BAFF and BAFF-R knockout (KO) mice cannot be used to investigate the requirements of IgA induction because they are deficient in B cells.39 TACI KO mice have low IgA and show poor immune responses to repetitive (type 2) T-independent antigens.40 In humans, mutations in the gene encoding TACI have been found in both common variable immunodeficient and IgA-deficient individuals.41, 42 In regard to APRIL, two APRIL-deficient mouse strains have been independently generated and they display either normal serum IgA levels43 or a selective decrease of IgA.44 The latter IgA-low APRIL-deficient mice also show defective IgA responses to mucosal immunization.44
In vitro, APRIL stimulates murine B cells to undergo CSR to IgG, IgA, and IgE via TACI, whereas TACI-deficient B cells switch to IgG and IgE, but not IgA when stimulated by BAFF.35 Taken together, APRIL–TACI interactions appear to be necessary for IgA production in vivo, and potentially make CD40-CD40L requirements redundant for IgA CSR.
If both APRIL and BAFF can stimulate CSR to isotypes other than IgA, then why is the IgA class switch so specific for the intestine and other mucosal sites? Intestinal dendritic cells from Peyer's patches and mesenteric lymph nodes constitutively secrete retinoic acid, which synergizes with IL-6 and IL-5 to induce IgA production in purified B cells, both in humans and in mice.45 Retinoic acid secretion alone can also induce the small intestinal homing receptor CCR9 on purified B cells.45 Thus, the environment of cytokines and dendritic cell phenotype in the intestinal lymphoid tissues is sufficient for the IgA-dominated response that is generated. In addition, intestinal epithelial cells may also influence the switch to IgA by the local secretion of mediators, such as BAFF and APRIL, TGFβ, and IL-6.
Germ-free mice, lacking the stimulus of the intestinal microbiota to IgA production, have a profound deficiency in IgA production in the intestinal mucosa, but the serum IgA levels are maintained at about half the normal magnitude.33 This suggests that some serum IgA (possibly mainly natural antibodies) can be produced independently of mucosal IgA. Another experiment that suggests separate circuits of systemic and mucosal IgA production has been carried out with alymphoblastic mice with no mucosal inductive sites other than tiny collections of nasal lymphoid tissue. In this strain, it is possible to reconstitute about half the serum IgA by adoptive transfer of wild-type B cells, but mucosal IgA-producing cells remain absent46 because the strain also has a defect in generating intestinal homing signals.47, 48
In addition to Peyer's patches, what other sources of IgA+ B cells do we have? In mice, both peritoneal B cells of the B1 lineage and marginal zone B cells in the spleen can contribute to IgA production (discussed further in the section Selective epithelial export of locally produced IgA and IgM). Since APRIL deficiency and TGFβRII deficiency lead to global lack of IgA, it is likely that these signaling mechanisms are also required for serum IgA production. Indeed, BAFF-transgenic mice show increased IgA switching in the marginal zone.49 In both purified B1 and marginal zone populations, culture with BAFF, LPS, and TGFβ stimulates the switch to IgA. The IgA switch is further enhanced in the case of marginal zone populations by the addition of anti-IgD linked to dextran, IL-4, and IL-5, whereas these cytokines do not increase the IgA switch in peritoneal B1 populations.50 These findings suggest that the requirements for CSR to IgA may be somewhat different at different sites. Whether retinoic acid or extra-intestinal dendritic cells are implicated in this pathway is currently unknown.
Immune Geography Of IgA Induction and Function
The above argument suggests that IgA can be induced at different tissue sites and from various subsets of B cells. We shall first discuss the classical pathway in intestinal lymphoid follicles.
The classical IgA+ B-cell Odyssey from intestinal lymphoid tissue
The distinct structure and function of IgA in mucosal immunity requires special immune geography to separate the inductive and effector arms so that they can function independently of the remainder of the immune system. The Odyssey of IgA induction and mucosal B-cell circulation is intrinsic to this immune geography, and consists of four stages: IgA induction in mucosal B cells; IgA+ plasmablast recirculation and homing to the intestinal mucosa; terminal B-cell differentiation to plasma cells with local IgA production; and export of IgA through the intestinal epithelial cell layer (see Figure 1 in the accompanying article by Brandtzaeg et al. in this issue, page XX).
The principal site of induction of the IgA was demonstrated by Craig and Cebra51 in 1971. In adoptive transfer experiments with Ig allotypic markers, they found that B cells from the Peyer's patches in rabbits switched to IgA. This showed that Peyer's patch B cells are destined to undergo CSR to IgA and to seed mucosal sites with precursors for IgA-producing plasma cells, despite the fact that few cells with cytoplasmic IgA are seen in the Peyer's patches. Much of the spontaneous IgA response in mice is driven by commensal intestinal bacteria, and Peyer's patch dendritic cells that have sampled commensals are responsible for the local IgA induction.52 It is now known for both humans and mice that intestinal dendritic cells constitutively secrete retinoic acid, and these program B cells both to undergo CSR to the α segment and also express the CCR9 molecule required for homing to the small intestine.45
Gowans et al.53 had shown in the 1950s and 1960s that lymphocytes generally home from the bloodstream to lymph nodes and then recirculate via efferent lymph back to the bloodstream at the junction of the thoracic duct and the left subclavian vein. Subsequently, they studied recirculation of IgA+ blasts induced in the gut.54 Using cholera toxin as an intestinal immunogen, they found that lymphoblasts secreting anti-cholera toxin IgA could be isolated from the thoracic duct lymph 16–19 days later, and that IgA-secreting cells were detected in the intestinal lamina propria from day 17. If two segments of the small intestine were surgically isolated in rats so that one part could be immunized with cholera toxin in isolation, recirculation nevertheless resulted in IgA-secreting cells reaching the lamina propria of an intestinal segment that had never been in contact with the immunogen.55
This B-cell homing system works through specific signals that identify the endothelial cells of the microvasculature in the intestinal lamina propria. In the systemic immune system, lymphocytes that are destined to leave the bloodstream interact with the vascular endothelium of high endothelial venules by tethering and rolling through interactions between their L-selectin (CD62L) and peripheral lymph node addressins, collectively called PNad. Firm arrest is mediated by chemokine-triggered integrin activation, and extravasation of cells then occurs through interaction of LFA-1 (αLβ2 integrin) with the generally expressed endothelial ICAM-1. At mucosal effector sites, vascular adhesion mechanisms differ from the systemic PNAd–CD62L interactions (reviewed in refs. 56, 57). Thus, adhesion is mediated mainly by chemokine-triggered integrin activation through α4β7 interacting with the mucosal addressin molecule MAdCAM-1, with some additional effect of αLβ2 affinity to endothelial ICAM-1. Moreover, tissue-specific expression of chemokine and chemokine receptors on memory/effector cells contributes to their preferential mucosal localization and retention (Figure 3). For example, IgA plasmablasts destined to home to the small intestine express CCR9 that binds to the locally produced ligand CCL25 (TECK), whereas large intestinal IgA plasmablasts express CCR10 and are attracted by the ligand CCL28 (MEC).

B-cell homing to mucosal and peripheral tissues is mediated by specific combinations of chemokine receptors and adhesion molecules. Newly developed B cells express specific chemokine receptors that mediate responsiveness to chemokine ligands expressed in secondary lymphoid tissues, bone marrow (BM), and mucosal effector sites. Within secondary lymphoid tissues, B cells encounter specific antigen leading to their activation and differentiation. In general, activation of B cells and differentiation to mucosal IgA antibody-secreting cells (plasmablasts and plasma cells) induce upregulation of CCR10 and expression of α4β1 integrin, which mediate attraction to CCL28 and VCAM-1, respectively. Expression of VCAM-1 in effector sites is indicated, although the expression of vascular adhesion molecules (like VCAM-1) at various effector sites is not yet systematically worked out. Differentiation of IgA plasmablasts within small intestinal lymphoid tissues additionally induces upregulation of CCR9 and α4β7 and directs homing back to the small intestine, which expresses CCL25 and MAdCAM-1. In contrast, differentiation to IgG plasmablasts of systemic type leads to upregulation of CXCR3, allowing responsiveness to CXCL9 and CXCL10 and migration to inflamed mucosal and non-mucosal tissues. Homing to the bone marrow is mediated through CXCR4, which is expressed on all types of plasmablasts, as well as CCR10. Memory B cells (not shown) retain expression of CCR7, CXCR5, and CXCR4 to allow their recirculation through lymphoid tissues. Retained expression of CCR7 and CD62L on effector B cells from tonsils/NALT may explain their joint tropism for organized lymphoid tissue and the upper aerodigestive tract (not shown). Specific chemokine–chemokine receptor pairs are indicated by corresponding colored text; specific integrin–ligand pairs are depicted by gray and yellow boxes. Bold text indicates the molecule is expressed on lymphocytes; text in italics indicates expression on endothelial or stromal cells. BALT, bronchus-associated lymphoid tissue; CCL, CC-chemokine ligand; CCR, CC-chemokine receptor; CXC-chemokine ligand; CXCL, VCAM-1, vascular cell-adhesion molecule-1; CXCR, CXC-chemokine receptor; MAdCAM-1, mucosal addressin cell-adhesion molecule-1; MLN, mesenteric lymph nodes; NALT, nasopharynx-associated lymphoid tissue; PP, Peyer's patches; PLN, peripheral lymph nodes.
Shared expression of adhesion and chemokine receptor pairs explains the fact that IgA lymphocytes induced in one mucosal site can populate another site with IgA-secreting cells, which gave rise to the obsolete term “a common mucosal immune system.” However, it is important to be aware of the extensive regionalization and compartmentalization that exist within the mucosal immune system.18, 58 Thus, in contrast to extravasation events in the intestine, α4β7 and MAdCAM-1 are not important homing molecules in the airways, where CD62L, α4β1, CCR7, and CCR10 appear to play a more decisive role (Figure 3).
Selective epithelial export of locally produced IgA and IgM
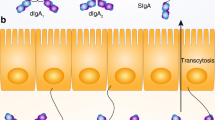
Most IgA+ mucosal plasma cells express the J chain and therefore produce dimers and larger polymers of IgA, collectively termed pIgA.18 The short J-chain polypeptide was identified independently by Koshland and co-workers59 and Mestecky et al.60 as an Ig-associated peptide common to SIgA and IgM. J-chain-mediated polymerization provides pIgA with enhanced antigen avidity, a property particularly useful for agglutinating microbes in external secretions. Furthermore, its selective incorporation into pIgA and pentameric IgM is mandatory for their binding to epithelial membrane SC, now best known as the pIgR.7, 61 This receptor carries its ligands to mucosal surfaces by a vesicular transport process termed transcytosis. Intracellular trafficking of pIgR has been studied in great detail and represents the paradigm for transcytosis across a polarized epithelium.62, 63
The pIgR has an N-terminal extracellular ligand-binding domain, a single membrane-spanning α helix, and a 103-amino-acid cytoplasmic C-terminal tail that contains all the information for proper intracellular routing.62 The extracellular region is comprised of five Ig-like domains (D1–D5). Solution structure analysis has suggested a bend between D3 and D4, forming a molecule reminiscent of a “J” separated from the transmembrane domain by a flexible region, which also contains the receptor cleavage site.64 The pIgR is a sacrificial transport receptor in that it is specifically cleaved to release its cargo. For this reason, it is constitutively expressed by mucosal epithelial cells at quite high levels, particularly in the small and large intestine.65 Furthermore, its expression can be regulated at the transcriptional level by cytokines and other mediators. Although one of us originally suggested that SC represented a transmembrane receptor,6 this was not formally proven until after the molecular cloning of rabbit pIgR by Mostov et al.66 in 1984. Its functional expression by transfection into Madin–Darby canine kidney (MDCK) cells as a transcytotic receptor for pIgA was demonstrated only 2 years later.67 The human pIgR was subsequently cloned,68 and similar transfection and transcytosis experiments have been made with this receptor.65
The pIgR is synthesized in the endoplasmic reticulum of secretory epithelial cells and delivered via the trans-Golgi network directly to the basolateral membrane. Here, it is available to bind J-chain-containing pIgA and pentameric IgM produced by lamina propria plasma cells.18 Binding is initiated by interactions between D1 of the receptor and the Ig Fc portion, and for IgA this non-covalent complexing is subsequently stabilized by covalent linking between D5 and Cα2.61 Receptor-mediated endocytosis of the ligand–pIgR complex, and also the unoccupied receptor, occurs by clathrin-mediated endocytosis. While some recycling back to the basolateral membrane occurs, the bulk of the receptor is ferried to the common endosome where cargo destined for transcytosis is segregated from cargo destined for basolateral recycling (such as the transferrin receptor).
The ligand–pIgR complex is next delivered to the apical recycling endosome and released to the intestinal lumen after fusion of the apical recycling endosome with the apical plasma membrane and cleavage of the pIgR by a leupeptin-sensitive endoprotease.69 Thus, cleavage of ligand-bound pIgR gives rise to SIg with bound SC, while cleavage of unoccupied pIgR gives rise to free SC. The covalent coupling of SC provides mucophilic properties and increased stability to SIgA in the harsh intestinal environment.70, 71 Owing to the lack of covalent stabilization by bound SC, SIgM is not as resistant as SIgA to degradation in the secretions. Despite this, SIgM can provide compensatory mucosal defence in early infancy and selective IgA deficiency.13 SIgM compensation is less consistent in the airways than in the gut, and this variable apparently contributes to susceptibility of IgA-deficient individuals to infections in the respiratory tract.72
The contribution of different tissue sites to IgA induction
As predicted by the classical Cebra paper,51 induction of mucosal IgA takes place largely in the organized gut-associated lymphoid tissue. For example, in isolated intestinal loops, cholera toxin immunization was ineffective in segments lacking a Peyer's patch.55 Other ways of looking at this effect are to exploit animals in which lymphoid structures are abnormal as a result of genetic manipulation or experimental disruption of the signaling mechanisms operative in secondary lymphoid tissue formation.
Peyer's patch organogenesis requires antenatal lymphotoxin (LT) α2β signaling between the CD4+CD3− organizer lymphoid cell population and the ICAM−1+ VCAM−1+ mesenchymal cell population around embryonic day e16.73 Mice deficient for LTα,74 LTβ,75 or LTβ receptor (LTβR) lack lymph nodes and Peyer's patches, and do not have intestinal IgA production. When LTβR signaling is disrupted by administration of the fusion protein of LTβR-Ig in utero, the offspring of mice are later still able to produce significant antigen-specific IgA responses following immunization.76 A more severe defect occurs if both Peyer's patches and mesenteric lymph nodes are disrupted by concurrent antenatal administration of LTβR-Ig and the TNFR55-Ig fusion proteins against the p55 subunit of the TNF receptor on days e13 and e16;77 in this case, antigen-specific responses are abrogated, although the total intestinal IgA is not significantly altered.
Peyer's patches are not the only gut-associated lymphoid tissue structures containing B lymphocytes in the gut wall; there are additional multiple solitary or isolated lymphoid follicles (ILFs)78 with a similar cellular composition. Like Peyer's patches, ILFs increase in cellularity during recolonization of germ-free mice.79 These structures also undergo a compensatory increase when Peyer's patch and mesenteric lymph node formation is disrupted by antenatal LTβR-Ig and TNFR55-Ig fusion protein treatment as described above. IgA induction in the germinal centers of ILFs probably accounts for the relatively normal intestinal IgA content under these conditions.77 The ILFs can be selectively disrupted by weekly administration of LTβR-Ig and TNFR55-Ig in young adult mice; in this case, the number of IgA-producing plasma cells in the intestine remains unaltered, but the response to intestinal immunization with tetanus toxoid is reduced.77
The conclusions from these studies are that both Peyer's patches and ILFs contribute to the generation of IgA-producing plasma cells in the intestinal mucosa. However, the absence of such plasma cells in mice with a genetic defect of LT signaling does not preclude extra-intestinal sites of IgA induction. Lymphoid tissue chemokines and adhesion molecules are reduced within the lamina propria of these mice, but their intestinal IgA deficiency can be partially reversed in a segment of transplanted RAG−/− intestine.48 Notably, in mice, both peritoneal B1 lymphocytes and splenic marginal zone B cells can contribute to IgA.
In embryonic and early neonatal life, B cells of mice are derived from the fetal liver, but lymphopoiesis is taken over by the bone marrow (B2 lymphocytes) and cells in the pleuroperitoneal cavities (B1 lymphocytes) after birth.80 B1 and B2 cells carry different B-cell receptor repertoires,81 can be distinguished by their surface markers, and apparently represent separate lineages.82, 83 Although there is no surface expression of IgA in peritoneal B-cell populations, experiments in which radiation chimeras have been made with allotypic markers to distinguish the IgA derived from B1 and B2 cells indicate that up to 50% of intestinal IgA (mostly T-independent) is B1-derived.33, 84, 85 In a different experimental system, intestinal IgA in MHC class II-deficient animals has been studied. Here, the levels of intestinal IgA were relatively normal, despite the T-cell deficiency and the absence of cognate B–T interactions; however, when the MHCII−/− strain also carried the xid mutation resulting in B-cell deficiency, IgA was severely reduced.
This is still a controversial area; experiments with antibody depletion followed by transfer of allotypically distinguishable peritoneal cells showed only little contribution of B1 cells to intestinal IgA,86 although residual recipient B1 cells in this system might have led to an underestimation. It is striking that both B1- and B2-derived IgA B-cell receptor repertoires are quite oligoclonal, without evidence of sequential acquisition of somatic mutations that would be expected in a conventional germinal center response. Both lineages may therefore function to generate a rather primitive B-cell response, probably as a high-capacity, low-affinity system directed against commensal bacterial surface epitopes.87
In general, these transfer or strain combination experiments leave uncertainty about the actual sites of CSR outside of gut-associated lymphoid tissue in vivo. A direct approach is hampered because the putative sites for CSR have only a few IgA+ B cells and because the nested PCR that detects the nascent transcripts from the circular DNA looped out between the switch regions on the Ig heavy-chain locus20 is a very demanding assay. Evidence for37, 88 and against22, 89 lamina propria CSR has been presented, and in the CD40-deficient strain, direct evidence of CSR was also not found in ILFs.22 B cells with B1 surface characteristics can be seen in Peyer's patches of TGFβRII−/− mice,26 and there is some indirect evidence that B1 CSR may occur in the mesenteric lymph nodes.90 Nevertheless, we still need much better information about the sites and mechanisms of alternative IgA CSR pathways.
Function of IgA In Different Systems
Non-pathogens
The lower intestine is inhabited by a dense commensal microbiota consisting of organisms that are not normally pathogenic for the host. Their numbers are prodigious: in humans, it is estimated that the number of intestinal bacteria (1014) outstrips the number of human cells in the body (1013) by an order of magnitude. The density of bacteria in the intestinal lumen (1012 g−1) is far higher than other microbial consortia found in soils, oceans, and other global subsurfaces (<108). As the best of good culture media, the human gut has to be highly adapted to the presence of these organisms and their potentially immunostimulatory molecules.
IgA is highly induced in the gut only in animals containing intestinal microbes; in those kept in a germ-free state, the number of intestinal IgA-producing plasma cells is 1–2 orders of magnitude lower. From this fact, teleology would suggest that IgA functions to protect the host. Although this can be shown in some cases for pathogens, the functional impact of SIgA is not easy to demonstrate for commensals, although they are probably responsible for most IgA induction. The reason is that 200 million years of mammalian evolution have endowed us with a mucosal immune system with a series of redundant protective mechanisms that allow us a mutual existence with our microbiota.91 These mechanisms are very diverse and involve systemic and mucosal immunity, epithelial biology, and metabolic mechanisms. In particular, IgA deficiency can be compensated in mouse and human by increased export of SIgM, which is also handled by the pIgR (see the section Selective epithelial export of locally produced IgA and IgM).
Two laboratories have made KO mice deficient for pIgR.92, 93 In both cases, there is a failure of SIgA and SIgM export into the intestinal lumen and highly increased concentrations of serum IgA. Serum IgG is also elevated and there is increased mucosal penetration of commensal bacteria and intensified priming of systemic antibody responses against these organisms.92, 94 Macpherson and Uhr52 showed that secretion of intestinal IgA limited penetration of test doses of intestinal commensals to the mesenteric lymph nodes. Johansen et al.92 also looked at the functional consequences of a system lacking SIg, and found serum protein loss into the intestinal lumen, pathognomonic of low-grade enteropathy. These results imply reduced mucosal protection against the commensal microbiota in the absence of secretory antibodies.
The SIgA protection mechanism is far less clear. Dimeric IgA has an abnormally large hydraulic diameter in relation to its molecular weight because of the combination of two Ig molecules;95 this may limit low-level penetration of IgA-coated microbes through the surface epithelium, or decrease the effective paracellular permeability of bound bacterial breakdown components. Furthermore, glycosylation of both IgA and the bound SC in SIgA may contribute to trapping of SIgA-bound antigen in the mucus.70, 71 A model for this is sperm penetration in cervical mucus; sperms coated with IgA show reduced motility and this effect is reversed by IgA1-specific protease.96
If microbes succeed in reaching and penetrating the surface epithelium, the SIgA export mechanism can act as a clearance system from the basolateral surface back to the lumen. Receptor-mediated epithelial IgA transport per se is clearly important in limiting epithelial viral infections in cell monolayers infected with Sendai97 and in vivo during rotavirus infection.17 In vivo measurements to assess the commensal penetration across the intestinal epithelium are currently mostly indirect and rely on bacterial cultivation from mesenteric lymph nodes. Results in adult wild-type mice and pIgR KO mice are described above. In neonatal mice, lack of maternal antibody in milk leads to premature induction of the pups’ own mucosal IgA from post-natal day 16 rather than after weaning at day 21.98 This results from stimulation by the commensal intestinal microbiota, because such early endogenous IgA induction in the pups does not occur under germ-free conditions.99 In challenge experiments in the pups, fixing the IgA specificity to nitrophenol with the quasimonoclonal mouse strain did not affect the ability of SIgA to limit penetration of the commensal Enterobacter cloacae.99 In other words, low antibody affinities to redundant surface epitopes of bacteria, or binding of IgA through bacterial lectin-mediated mechanisms, can probably be sufficient for the reinforced barrier effect of SIgA.
The simplistic notion of an immunological ‘fly-paper’ mechanism on the luminal side of the epithelium could also be valid, as SIgA may increase the barrier by stabilizing a biofilm layer of bacteria. For example, binding of purified salivary mucin to Pseudomonas aeruginosa and Staphylococcus aureus depends on SIgA because the binding disappears when SIgA is removed, but can be reconstituted when it is reintroduced.100 Moreover, formation of bacterial biofilms in culture on glutaraldehyde-fixed CaCo2 cells can be promoted by SIgA and mucin, and remain stable despite repetitive replacement of the medium.101 Formation of biofilms dependent on pili expression has also been shown to be SIgA-dependent.102
Paradoxically, the protective mechanisms argued to apply at the level of the surface epithelium may work in the opposite direction in the M cells of the gut-associated lymphoid tissue. Here uptake of SIgA, complexed with its antigen, can increase the sampling of intestinal bacteria.103
Microbial pathogens
The functional results of experiments with pathogens at mucosal surfaces have also shown a role for SIgA protection. Because of its remarkable immunostimulatory properties, cholera toxin has been used as a potent mucosal adjuvant.104, 105, 106 However, its toxicity prohibits application in its native form in humans. Injection of a test dose of cholera toxin into ligated intestinal loops causes great accumulation of fluid, and prior mucosal immunization can generate toxin-neutralizing SIgA antibody in vivo.107 Unlike SIgA protection against the commensal microflora, this does require the conventional mechanisms of T-cell help for a germinal center response, presumably to raise the antibody affinity to satisfactory functional levels, as well as active pIgR-mediated epithelial transport.14, 29, 108, 109, 110 Orally immunized J-chain and pIgR KO mice are as susceptible to cholera toxin as naive mice.111 In the naive state, mice lacking secretory antibodies are more susceptible to infection with Salmonella typhimurium than wild-type animals.112 Thus, natural SIgA antibodies inhibited early invasion and horizontal fecal-oral spread of this pathogen.
Influenza infection in mice is also abrogated by SIgA-neutralizing antibodies to the virus. Adoptive transfer of doses of antibodies from immune animals of different isotypes matched to those in convalescent serum shows that SIgA prevents pathology in the upper respiratory tract whereas IgG prevents pathology in the peripheral lung parenchyma.113
Similarly, where mice have been immunized with the H1N1 vaccine and challenged 4–5 weeks later with 103 PFU of the A/PR/8/34 strain intranasally, IgA-deficient mice showed increased lethality, which could be abrogated if the immunization was accompanied by the addition of IL-12.114 In this system, SIgA was one of a number of protective mechanisms, and IL-12 was shown to correct the poor T-cell proliferative responses to phytohemoagglutinin and memory recall responses to H1N1 in IgAKo animals. Not surprisingly, selective IgA deficiency in an otherwise unmanipulated animal does not significantly alter the outcome of influenza infection in experiments based on a strong adjuvant.115 However, cross-protection against antigenically distinct, but related A-type strains of influenza virus was abrogated in pIgR KO mice, suggesting a protecting role of crossreactive SIgA antibodies.116
In experimental murine rotavirus infection, clearance of virus correlated well with production of mucosal and serum IgA,117, 118 and specific IgA was induced even in T-cell-deficient animals.119 Although B cells were required to protect mice from experimental re-infection with virus,120, 121 experiments with the IgA KO strain showed that SIgA is potentially redundant for the protective effect.122 Interestingly, although from observations that J chain KO mice showed delayed clearance of rotavirus and failure of immunization123 one would expect that SIgM should compensate for IgA in the IgAKo strain, it appeared to be IgG rather than IgM that provided protection with both serum and fecal antibodies.122
This picture of IgA occasionally having a redundant role in pathogen protection at mucosal surfaces with compensation by antibodies of other isotypes, or by innate immune mechanisms, is a general one. For example, experimental vaginal infection with HSV2 in IgA KO mice appears not to be different from that in wild-type strains,124, 125 and IgA is also not essential for genital tract chlamydia immunity in mice.126 Nevertheless, clearance of intestinal Giardia muris infection was abrogated in IgA KO, pIgR KO, and J chain KO mice, indicating an important role for SIgA in expulsion of this lumen-dwelling parasite.127
Epilogue
IgA is an appropriate subject of a review in the first issue of this journal because its history reaches back to the start of mucosal immunology, and its immune geography is inextricable from the distinct nature of the mucosal immune system. Indeed, the 1st International Congress of Mucosal Immunology, held in Birmingham (AL, USA) in 1973, was called a symposium on the immunoglobulin A system (organized by Frederick W. Kraus, Jiri Mestecky, and Alexander R. Lawton). Yet compared with the dominance of IgA in total body Ig production, relatively little is known about its function, and especially its role in host–microbial mutualism remains elusive. This is a paradigm for mucosal immunology: a major part of immunity that is relatively understudied and where more information is badly needed. The new journal aims to be a valuable vehicle to stimulate and disseminate the results of such studies.
Disclosure
The authors declared no conflict of interest.
References
Gugler, E., Bokelmann, G., Datwyler, A. & Von Muralt, G. [Immunoelectrophoretic studies on human milk proteins]. Schweiz. Med. Wochenschr. 88, 1264–1267 (1958).
Heremans, J.F., Heremans, M.T. & Schultze, H.E. Isolation and description of a few properties of the beta 2A-globulin of human serum. Clin. Chim. Acta 4, 96–102 (1959).
Chodirker, W.B. & Tomasi, T.B. Jr Gamma-globulins: quantitative relationships in human serum and nonvascular fluids. Science 142, 1080–1081 (1963).
Hanson, L.A. & Brandtzaeg, P. The discovery of secretory IgA and the mucosal immune system. Immunol. Today 14, 416–417 (1993).
Tomasi, T.B. Jr, Tan, E.M., Solomon, A. & Prendergast, R.A. Characteristics of an immune system common to certain external secretions. J. Exp. Med. 121, 101–124 (1965).
Brandtzaeg, P. Mucosal and glandular distribution of immunoglobulin components: differential localization of free and bound SC in secretory epithelial cells. J. Immunol. 112, 1553–1559 (1974).
Brandtzaeg, P. & Prydz, H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature 311, 71–73 (1984).
Tomasi, T.B. Jr & Bienenstock, J. Secretory immunoglobulins. Adv. Immunol. 9, 1–96 (1968).
Whitman, W.B., Coleman, D.C. & Wiebe, W.J. Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. USA 95, 6578–6583 (1998).
Benveniste, J., Lespinats, G. & Salomon, J. Serum and secretory IgA in axenic and holoxenic mice. J. Immunol. 107, 1656–1662 (1971).
Benveniste, J., Lespinats, G., Adam, C. & Salomon, J.C. Immunoglobulins in intact, immunized, and contaminated axenic mice: study of serum IgA. J. Immunol. 107, 1647–1655 (1971).
Moreau, M.C., Ducluzeau, R., Guy-Grand, D. & Muller, M.C. Increase in the population of duodenal immunoglobulin A plasmocytes in axenic mice associated with different living or dead bacterial strains of intestinal origin. Infect. Immun. 21, 532–539 (1978).
Brandtzaeg, P., Nilssen, D.E., Rognum, T.O. & Thrane, P.S. Ontogeny of the mucosal immune system and IgA deficiency. Gastroenterol. Clin. N. Am. 20, 397–439 (1991).
Lycke, N., Eriksen, L. & Holmgren, J. Protection against cholera toxin after oral immunisation is thymus dependent and associated with intestinal production of neutralising IgA antitoxin. Scand. J. Immunol. 25, 413–419 (1987).
Kalinke, U., Bucher, EM., Ernst, B., Oxenius, A., Roost, HP., Geley, S. et al. The role of somatic mutation in the generation of the protective humoral immune response against vesicular stomatitis virus. Immunity 5, 639–652 (1996).
Kaetzel, C.S., Robinson, J.K., Chintalacharuvu, K.R., Vaerman, J.P. & Lamm, M.E. The polymeric immunoglobulin receptor (secretory component) mediates transport of immune complexes across epithelial cells: a local defense function for IgA. Proc. Natl. Acad. Sci. USA 88, 8796–8800 (1991).
Burns, J.W., Siadat-Pajouh, M., Krishnaney, A.A. & Greenberg, H.B. Protective effect of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science 272, 104–107 (1996).
Brandtzaeg, P. & Johansen, F.E. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol. Rev. 206, 32–63 (2005).
Chaudhuri, J. & Alt, F.W. Class-switch recombination: interplay of transcription DNA deamination and DNA repair. Nat. Rev. Immunol. 4, 541–552 (2004).
Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y. & Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563 (2000).
Kingzette, M., Spieker-Polet, H., Yam, P.C., Zhai, S.K. & Knight, K.L. Trans-chromosomal recombination within the Ig heavy chain switch region in B lymphocytes. Proc. Natl. Acad. Sci. USA 95, 11840–11845 (1998).
Bergqvist, P., Gardby, E., Stensson, A., Bemark, M. & Lycke, N.Y. Gut IgA class switch recombination in the absence of CD40 does not occur in the lamina propria and is independent of germinal centers. J. Immunol. 177, 7772–7783 (2006).
Renshaw, B.R., Fanslow, W.C. III, Armitage, R.J., Campbell, K.A., Liggitt, D., Wright, B. et al. Humoral immune responses in CD40 ligand-deficient mice. J. Exp. Med. 180, 1889–1900 (1994).
Sonoda, E., Hitoshi, Y., Yamaguchi, N., Ishii, T., Tominaga, A., Araki, S. et al. Differential regulation of IgA production by TGF-beta and IL-5: TGF-beta induces surface IgA-positive cells bearing IL-5 receptor, whereas IL-5 promotes their survival and maturation into IgA-secreting cells. Cell Immunol. 140, 158–172 (1992).
Macpherson, A.J., Hunziker, L., McCoy, K. & Lamarre, A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 3, 1021–1035 (2001).
Cazac, B.B. & Roes, J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 13, 443–451 (2000).
Li, R., Rosendahl, A., Brodin, G., Cheng, A.M., Ahgren, A., Sundquist, C. et al. Deletion of exon I of SMAD7 in mice results in altered B cell responses. J. Immunol. 176, 6777–6784 (2006).
Klein, J., Ju, W., Heyer, J., Wittek, B., Haneke, T., Knaus, P. et al. B cell-specific deficiency for Smad2 in vivo leads to defects in TGF-beta-directed IgA switching and changes in B cell fate. J. Immunol. 176, 2389–2396 (2006).
Vajdy, M., Kosco-Vilbois, M.H., Kopf, M., Kohler, G. & Lycke, N. Impaired mucosal immune responses in interleukin 4-targeted mice. J. Exp. Med. 181, 41–53 (1995).
Kopf, M., Le Gros, G., Coyle, A.J., Kosco-Vilbois, M. & Brombacher, F. Immune responses of IL-4, IL-5, IL-6 deficient mice. Immunol. Rev. 148, 45–69 (1995).
Bromander, A.K., Ekman, L., Kopf, M., Nedrud, J.G. & Lycke, N.Y. IL-6-deficient mice exhibit normal mucosal IgA responses to local immunizations and Helicobacter felis infection. J. Immunol. 156, 4290–4297 (1996).
Ramsay, A.J., Husband, A.J., Ramshaw, I.A., Bao, S., Matthaei, K.I., Koehler, G. et al. The role of interleukin-6 in mucosal IgA antibody responses in vivo. Science 264, 561–563 (1994).
Macpherson, A.J., Gatto, D., Sainsbury, E., Harriman, G.R., Hengartner, H. & Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226 (2000).
Litinskiy, M.B., Nardelli, B., Hilbert, D.M., Schaffer, A., Casali, P. & Cerutti, A. Antigen presenting cells induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 3, 822–829 (2002).
Castigli, E., Wilson, S.A., Scott, S.,, Dedeoglu, F., Xu, S., Lam, K.P. et al. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 201, 35–39 (2005).
Mackay, F., Schneider, P., Rennert, P. & Browning, J. BAFF and APRIL: a tutorial on B cell survival. Annu. Rev. Immunol. 21, 231–264 (2003).
He, B., Xu, W., Santini, P.A., Polydorides, A.D., Chiu, A., Estrella, J. et al. Intestinal bacteria trigger T cell-independent Immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 26, 812–826 (2007).
Ingold, K., Zumsteg, A., Tardivel, A., Huard, B., Steiner, Q.G., Cachero, T.G. et al. Identification of proteoglycans as the APRIL-specific binding partners. J. Exp. Med. 201, 1375–1383 (2005).
Rolink, A.G. & Melchers, F. BAFFled B cells survive and thrive: roles of BAFF in B-cell development. Curr. Opin. Immunol. 14, 266–275 (2002).
von Bulow, G.U., van Deursen, J.M. & Bram, R.J. Regulation of the T-independent humoral response by TACI. Immunity 14, 573–582 (2001).
Castigli, E., Wilson, S.A., Garibyan, L., Rachid, R., Bonilla, F., Schneider, L. et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet. 37, 829–834 (2005).
Salzer, U., Chapel, H.M., Webster, A.D., Pan-Hammarstrom, Q., Schmitt-Graeff, A., Schlesier, M. et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat. Genet. 37, 820–828 (2005).
Varfolomeev, E., Kischkel, F., Martin, F., Seshasayee, D., Wang, H., Lawrence, D. et al. APRIL-deficient mice have normal immune system development. Mol. Cell. Biol. 24, 997–1006 (2004).
Castigli, E., Scott, S., Dedeoglu, F., Bryce, P., Jabara, H., Bhan, A.K. et al. Impaired IgA class switching in APRIL-deficient mice. Proc. Natl. Acad. Sci. USA 101, 3903–3908 (2004).
Mora, J.R., Iwata, M., Eksteen, B., Song, S.Y., Junt, T., Senman, B. et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006).
Macpherson, A.J., Lamarre, A., McCoy, K., Dougan, G., Harriman, G., Hengartner, H. et al. IgA B cell and IgA antibody production in the absence of mu and delta heavy chain expression early in B cell ontogeny. Nat. Immunol. 2, 625–631 (2001).
Fagarasan, S., Shinkura, R., Kamata, T., Nogaki, F., Ikuta, K., Tashiro, K. et al. Alymphoplasia (aly)-type nuclear factor kappaB-inducing kinase (NIK) causes defects in secondary lymphoid tissue chemokine receptor signaling and homing of peritoneal cells to the gut-associated lymphatic tissue system. J. Exp. Med. 191, 1477–1486 (2000).
Kang, H.S., Chin, R.K., Wang, Y., Yu, P., Wang, J., Newell, K.A. et al. Signaling via LTbetaR on the lamina propria stromal cells of the gut is required for IgA production. Nat. Immunol. 3, 576–582 (2002).
Batten, M. & Groom, J. BAFF mediates survival of peripheral immature B lymphocytes. J. Exp. Med. 192, 1453–1466 (2000).
Kaminski, D.A. & Stavnezer, J. Enhanced IgA class switching in marginal zone and B1 B cells relative to follicular/B2 B cells. J. Immunol. 177, 6025–6029 (2006).
Craig, S.W. & Cebra, J.J. Peyer's patches: an enriched source of precursors for IgA-producing immunocytes in the rabbit. J. Exp. Med. 134, 188–200 (1971).
Macpherson, A.J. & Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 (2004).
Gowans, J.L. & Knight, E.J. The route of recirculation of lymphocytes in the rat. Proc. R. Soc. London B 159, 257–282 (1964).
Pierce, N.F. & Gowans, J.L. Cellular kinetics of the intestinal immune response to cholera toxoid in rats. J. Exp. Med. 142, 1550–1563 (1975).
Husband, A.J. & Gowans, J.L. The origin and antigen-dependent distribution of IgA-containing cells in the intestine. J. Exp. Med. 148, 1146–1160 (1978).
Kunkel, E.J. & Butcher, E.C. Plasma-cell homing. Nat. Rev. Immunol. 3, 822–829 (2003).
Kunkel, E.J. & Butcher, E.C. Chemokines and the tissue-specific migration of lymphocytes. Immunity 16, 1–4 (2002).
Johansen, F.E., Baekkevold, E.S., Carlsen, H.S., Farstad, I.N., Soler, D. & Brandtzaeg, P. Regional induction of adhesion molecules and chemokine receptors explains disparate homing of human B cells to systemic and mucosal effector sites: dispersion from tonsils. Blood 106, 593–600 (2005).
Halpern, M.S. & Koshland, M.E. Noval subunit in secretory IgA. Nature 228, 1276–1278 (1970).
Mestecky, J., Zikan, J. & Butler, W.T. Immunoglobulin M and secretory immunoglobulin A: presence of a common polypeptide chain different from light chains. Science 171, 1163–1165 (1971).
Johansen, F.E., Braathen, R. & Brandtzaeg, P. Role of J chain in secretory immunoglobulin formation. Scand. J. Immunol. 52, 240–248 (2000).
Mostov, K. & Kaetzel, C.S. Immunoglobulin transport and the polymeric immunoglobulin receptor. In Mucosal Immunology (Ogra, P.L., Mestecky, J., Lamm, M.E., Strober, W. Bienenstock, J. and McGhee, J.R., eds) 133 (Academic Press, San Diego, 1999).
Mostov, K., Su, T. & ter Beest, M. Polarized epithelial membrane traffic: conservation and plasticity. Nat. Cell Biol. 5, 287–293 (2003).
Bonner, A., Perrier, C., Corthesy, B. & Perkins, S.J. Solution structure of human secretory component: implications for biological function. J. Biol. Chem. 282, 16969–16980 (2007).
Johansen, F.E. & Brandtzaeg, P. Transcriptional regulation of the mucosal IgA system. Trends Immunol. 25, 150–157 (2004).
Mostov, K.E., Friedlander, M. & Blobel, G. The receptor for transepithelial transport of IgA and IgM contains multiple immunoglobulin-like domains. Nature 308, 37–43 (1984).
Mostov, K.E. & Deitcher, D.L. Polymeric immunoglobulin receptor expressed in MDCK cells transcytoses IgA. Cell 46, 613–621 (1986).
Krajci, P. Molecular cloning of the human transmembrane secretory component (poly-Ig receptor) and its mRNA expression in human tissues. Biochem. Biophys. Res. Commun. 158, 783–789 (1989).
Rojas, R. & Apodaca, G. Immunoglobulin transport across polarized epithelial cells. Nat. Rev. Mol. Cell Biol. 3, 944–955 (2002).
Crottet, P. & Corthesy, B. Secretory component delays the conversion of secretory IgA into antigen-binding competent F(ab′)2: a possible implication for mucosal defense. J. Immunol. 161, 5445–5453 (1998).
Phalipon, A. & Corthesy, B. Novel functions of the polymeric Ig receptor: well beyond transport of immunoglobulins. Trends Immunol. 24, 55–58 (2003).
Brandtzaeg, P. & Nilssen, D.E. Mucosal aspects of primary B cell deficiency and gastrointestinal infections. Curr. Opin. Gastroenterol. 11, 532–540 (1995).
Honda, K., Nakano, H., Yoshida, H., Nishikawa, S., Rennert, P., Ikuta, K. et al. Molecular basis for hematopoietic/mesenchymal interaction during initiation of Peyer's patch organogenesis. J. Exp. Med. 193, 621–630 (2001).
Banks, T.A., Rouse, B.T., Kerley, M.K., Blair, P.J., Godfrey, V.L., Kuklin, N.A. et al. Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J. Immunol. 155, 1685–1693 (1995).
Koni, P.A., Sacca, R., Lawton, P., Browning, J.L., Ruddle, N.H. & Flavell, R.A Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity 6, 491–500 (1997).
Yamamoto, M., Rennert, P., McGhee, J.R., Kweon, M.-N., Yamamoto, S., Dohi, T. et al. Alternate mucosal immune system: organized Peyer's patches are not required for IgA responses in the gastrointestinal tract. J. Immunol. 164, 5184–5191 (2000).
Yamamoto, M., Kweon, M.N., Rennert, P.D., Hiroi, T., Fujihashi, K., McGhee, J.R. et al. Role of gut-associated lymphoreticular tissues in antigen-specific intestinal IgA immunity. J. Immunol. 173, 762–769 (2004).
Hamada, H., Hiroi, T., Nishiyama, Y., Takahashi, H., Masunaga, Y., Hachimura, S. et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 168, 57–64 (2002).
Pabst, O., Herbrand, H., Friedrichsen, M., Velaga, S., Dorsch, M., Berhardt, G. et al. Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J. Immunol. 177, 6824–6832 (2006).
Hayakawa, K. & Hardy, R.R. Normal, autoimmune, and malignant CD5+ B cells: the Ly-1 B lineage? Annu. Rev. Immunol. 6, 197–218 (1988).
Lam, K.P. & Rajewsky, K. B cell antigen receptor specificity and surface density together determine B-1 versus B-2 cell development. J. Exp. Med. 190, 471–478 (1999).
Montecino-Rodriguez, E., Leathers, H. & Dorshkind, K. Identification of a B-1 B cell-specified progenitor. Nat. Immunol. 7, 293–301 (2006).
Tung, J.W., Mrazek, M.D., Yang, Y., Herzenberg, L.A. & Herzenberg, L.A. Phenotypically distinct B cell development pathways map to the three B cell lineages in the mouse. Proc. Natl. Acad. Sci. USA 103, 6293–6298 (2006).
Hayakawa, K., Hardy, R.R., Stall, A.M. & Herzenberg, L.A. Immunoglobulin-bearing B cells reconstitute and maintain the murine Ly-1 B cell lineage. Eur. J. Immunol. 16, 1313–1316 (1986).
Kroese, F.G., Butcher, E.C., Stall, A.M., Lalor, P.A., Adams, S. & Herzenberg, L.A. Many of the IgA producing plasma cells in murine gut are derived from self-replenishing precursors in the peritoneal cavity. Int. Immunol. 1, 75–84 (1989).
Thurnheer, M.C., Zuercher, A.W., Cebra, J.J. & Bos, N.A. B1 cells contribute to serum IgM but not to intestinal IgA production in gnotobiotic Ig allotype chimeric mice. J. Immunol. 170, 4564–4571 (2003).
Stoel, M., Jiang, H.-Q., van, Diemen C.C., Bun, J.C.A.M., Dammers, P.M., Thurnheer, M.C. et al. Restricted IgA repertoire in both B-1 and B-2 cell-derived gut plasmablasts. J. Immunol. 174, 1046–1054 (2005).
Fagarasan, S., Kinoshita, K., Muramatsu, M., Ikuta, K. & Honjo, T. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature 413, 639–643 (2001).
Shikina, T., Hiroi, T., Iwatani, K., Jang, M.H., Fukuyama, S., Tamura, M. et al. IgA class switch occurs in the organized nasopharynx- and gut-associated lymphoid tissue, but not in the diffuse lamina propria of airways and gut. J. Immunol. 172, 6259–6264 (2004).
Bos, N.A., Bun, J.C., Popma, S.H., Cebra, E.R., Deenen, G.J., van, der Cammen M.J. et al. Monoclonal immunoglobulin A derived from peritoneal B cells is encoded by both germ line and somatically mutated VH genes and is reactive with commensal bacteria. Infect. Immun. 64, 616–623 (1996).
Mueller, C. & Macpherson, A.J. Layers of mutualism with commensal bacteria protect us from intestinal inflammation. Gut 55, 276–284 (2006).
Johansen, F.E., Pekna, M., Norderhaug, I.N., Haneberg, B., Hietala, M.A., Krajci, P. et al. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 190, 915–922 (1999).
Shimada, S.-i., Kawaguchi-Miyashita, M., Kushiro, A., Sato, T., Nanno, M., Sako, T. et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J. Immunol. 163, 5367–5373 (1999).
Sait, L.C., Galic, M., Price, J.D., Simpfendorfer, K.R., Diavatopoulos, D.A., Uren, T.K. et al. Secretory antibodies reduce systemic antibody responses against the gastrointestinal commensal flora. Int. Immunol. 19, 257–265 (2007).
Saltzman, W.M., Radomsky, M.L., Whaley, K.J. & Cone, R.A. Antibody diffusion in human cervical mucus. Biophys. J. 66, 508–515 (1994).
Bronson, R.A., Cooper, G.W., Rosenfeld, D.L., Gilbert, J.V. & Plaut, A.G. The effect of an IgA1 protease on immunoglobulins bound to the sperm surface and sperm cervical mucus penetrating ability. Fertil. Steril. 47, 985–991 (1987).
Mazanec, M.B., Kaetzel, C.S., Lamm, M.E., Fletcher, D. & Nedrud, J.G. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc. Natl. Acad. Sci. USA 89, 6901–6905 (1992).
Kramer, D.R. & Cebra, J.J. Early appearance of “natural” mucosal IgA responses and germinal centers in suckling mice developing in the absence of maternal antibodies. J. Immunol. 154, 2051–2062 (1995).
Harris, N.L., Spoerri, I., Schopfer, J.F., Nembrini, C., Merky, P., Massacand, J. et al. Mechanisms of neonatal mucosal antibody protection. J. Immunol. 177, 6256–6262 (2006).
Biesbrock, A.R., Reddy, M.S. & Levine, M.J. Interaction of a salivary mucin-secretory immunoglobulin A complex with mucosal pathogens. Infect. Immun. 59, 3492–3497 (1991).
Bollinger, R.R., Everett, M.L., Palestrant, D., Love, S.D., Lin, S.S. & Parker, W. Human secretory immunoglobulin A may contribute to biofilm formation in the gut. Immunology 109, 580–587 (2003).
Bollinger, R.R., Everett, M.L., Wahl, S.D., Lee, Y.H., Orndorff, P.E. & Parker, W. Secretory IgA and mucin-mediated biofilm formation by environmental strains of Escherichia coli: role of type 1 pili. Mol. Immunol. 43, 378–387 (2006).
Corthesy, B. Roundtrip ticket for secretory IgA: role in mucosal homeostasis? J. Immunol. 178, 27–32 (2007).
Elson, C.O. & Ealding, W. Cholera toxin feeding did not induce oral tolerance in mice and abrogated oral tolerance to an unrelated protein antigen. J. Immunol. 133, 2892–2897 (1984).
Elson, C.O. & Ealding, W. Generalized systemic and mucosal immunity in mice after mucosal stimulation with cholera toxin. J. Immunol. 132, 2736–2741 (1984).
Elson, C.O. & Ealding, W. Genetic control of the murine immune response to cholera toxin. J. Immunol. 135, 930–932 (1985).
Lycke, N. & Holmgren, J. Intestinal mucosal memory and presence of memory cells in lamina propria and Peyer's patches in mice 2 years after oral immunization with cholera toxin. Scand. J. Immunol. 23, 611–616 (1986).
Hornquist, C., Ekman, L., Grdic, K., Schon, K. & Lycke, N. Paradoxical IgA immunity in CD4-deficient mice. Lack of cholera toxin-specific protective immunity despite normal gut mucosal IgA differentiation. J. Immunol. 155, 2877–2887 (1995).
Gardby, E., Lane, P. & Lycke, N.Y. Requirements for B7-CD28 costimulation in mucosal IgA responses: paradoxes observed in CTLA4-H gamma 1 transgenic mice. J. Immunol. 161, 49–59 (1998).
Lycke, N., Erlandsson, L., Ekman, L., Schon, K. & Leanderson, T. Lack of J chain inhibits the transport of gut IgA and abrogates the development of intestinal antitoxic protection. J. Immunol. 163, 913–919 (1999).
Uren, T.K., Wijburg, O.L., Simmons, C., Johansen, F.E., Brandtzaeg, P. & Strugnell, R.A. Vaccine-induced protection against gastrointestinal bacterial infections in the absence of secretory antibodies. Eur. J. Immunol. 35, 180–188 (2005).
Wijburg, O.L., Uren, T.K., Simpfendorfer, K., Johansen, F.E., Brandtzaeg, P. & Strugnell, R.A. Innate secretory antibodies protect against natural Salmonella typhimurium infection. J. Exp. Med. 203, 21–26 (2006).
Renegar, K.B., Small, P.A. Jr, Boykins, L.G. & Wright, P.F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 173, 1978–1986 (2004).
Arulanandam, B.P., Raeder, R.H., Nedrud, J.G., Bucher, D.J., Le, J. & Metzger, D.W. IgA immunodeficiency leads to inadequate Th cell priming and increased susceptibility to Influenza virus infection. J. Immunol. 166, 226–231 (2001).
Mbawuike, I.N., Pacheco, S., Acuna, C.L., Switzer, K.C., Zhang, Y.X. & Harriman, G.R. Mucosal immunity to influenza without IgA: an IgA knockout mouse model. J. Immunol. 162, 2530–2537 (1999).
Asahi, Y., Yoshikawa, T., Watanabe, I., Iwasaki, T., Hasegawa, H., Sato, Y. et al. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J. Immunol. 168, 2930–2938 (2002).
Burns, J.W., Krishnaney, A.A., Vo, P.T., Rouse, R.V., Anderson, L.J. & Greenberg, H.B. Analyses of homologous rotavirus infection in the mouse model. Virology 207, 143–153 (1995).
Feng, N., Burns, J.W., Bracy, L. & Greenberg, H.B. Comparison of mucosal and systemic humoral immune responses and subsequent protection in mice orally inoculated with a homologous or a heterologous rotavirus [published erratum appears in J Virol 1995 69(5):3246]. J. Virol. 68, 7766–7773 (1994).
Franco, M. & Greenberg, H.B. Immunity to rotavirus in T cell deficient mice. Virology 238, 169–179 (1997).
Franco, M.A. & Greenberg, H.B. Role of B cells and cytotoxic T lymphocytes in clearance of and immunity to rotavirus infection in mice. J. Virol. 69, 7800–7806 (1995).
McNeal, M.M., Stone, S.C., Basu, M., Bean, J.A., Clements, J.D., Hendrickson, B.A. et al. Protection against rotavirus shedding after intranasal immunization of mice with a chimeric VP6 protein does not require intestinal IgA. Virology 346, 338–347 (2006).
O’Neal, C.M., Harriman, G.R. & Conner, M.E. Protection of the villus epithelial cells of the small intestine from rotavirus infection does not require immunoglobulin A. J. Virol. 74, 4102–4109 (2000).
Schwartz-Cornil, I., Benureau, Y., Greenberg, H., Hendrickson, B.A. & Cohen, J. Heterologous protection induced by the inner capsid proteins of rotavirus requires transcytosis of mucosal immunoglobulins. J. Virol. 76, 8110–8117 (2002).
Parr, M.B., Harriman, G.R. & Parr, E.L. Immunity to vaginal HSV-2 infection in immunoglobulin A knockout mice. Immunology 95, 208–213 (1998).
Parr, E.L. & Parr, M.B. Immunoglobulin G is the main protective antibody in mouse vaginal secretions after vaginal immunization with attenuated herpes simplex virus type 2. J. Virol. 71, 8109–8115 (1997).
Morrison, S.G. & Morrison, R.P. The protective effect of antibody in immunity to murine chlamydial genital tract reinfection is independent of Immunoglobulin A. Infect. Immun. 73, 6183–6186 (2005).
Davids, B.J., Palm, J.E., Housley, M.P., Smith, J.R., Andersen, Y.S., Martin, M.G. et al. Polymeric immunoglobulin receptor in intestinal immune defense against the lumen-dwelling protozoan parasite Giardia. J. Immunol. 177, 6281–6290 (2006).
Furtado, P.B., Whitty, P.W., Robertson, A., Eaton, J.T., Almogren, A., Kerr, M.A. et al. Solution structure determination of monomeric human IgA2 by X-ray and neutron scattering, analytical ultracentrifugation and constrained modelling: a comparison with monomeric human IgA1. J. Mol. Biol. 338, 921–941 (2004).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Macpherson, A., McCoy, K., Johansen, FE. et al. The immune geography of IgA induction and function. Mucosal Immunol 1, 11–22 (2008). https://doi.org/10.1038/mi.2007.6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2007.6
This article is cited by
-
Tango of B cells with T cells in the making of secretory antibodies to gut bacteria
Nature Reviews Gastroenterology & Hepatology (2023)
-
Determination of soluble tumor necrosis factor receptor II and secretory immunoglobulin A in saliva of patients with dementia
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Recent progress in practical applications of a potential carotenoid astaxanthin in aquaculture industry: a review
Fish Physiology and Biochemistry (2023)
-
Issues for patchy tissues: defining roles for gut-associated lymphoid tissue in neurodevelopment and disease
Journal of Neural Transmission (2023)
-
Role of FcγRIII in the nasal cavity of BALB/c mice in the primary amebic meningoencephalitis protection model
Parasitology Research (2023)
