
Abstract
Blinatumomab can induce a complete haematological remission in patients in 46.6% with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL) resulting in a survival benefit when compared with chemotherapy. Only bone marrow blast counts before therapy have shown a weak prediction of response. Here we investigated the role of regulatory T cells (Tregs), measured by CD4/CD25/FOXP3 expression, in predicting the outcome of immunotherapy with the CD19-directed bispecific T-cell engager construct blinatumomab. Blinatumomab responders (n=22) had an average of 4.82% Tregs (confidence interval (CI): 1.79–8.34%) in the peripheral blood, whereas non-responders (n=20) demonstrated 10.25% Tregs (CI: 3.36–65.9%). All other tested markers showed either no prediction value or an inferior prediction level including blast BM counts and the classical enzyme marker lactate dehydrogenase. With a cutoff of 8.525%, Treg enumeration can identify 100% of all blinatumomab responders and exclude 70% of the non-responders. The effect is facilitated by blinatumomab-activated Tregs, leading to interleukin-10 production, resulting in suppression of T-cell proliferation and reduced CD8-mediated lysis of ALL cells. Proliferation of patients' T cells can be restored by upfront removal of Tregs. Thus, enumeration of Treg identifies r/r ALL patients with a high response rate to blinatumomab. Therapeutic removal of Tregs may convert blinatumomab non-responders to responders.
Similar content being viewed by others


Introduction
Adult patients with relapsed or refractory acute lymphoblastic leukaemia (r/r ALL) have a poor prognosis despite intense salvage chemotherapy following an allogeneic hematopoetic stem cell transplant. Overall, the response rate to salvage chemotherapy is only 30–45% and the median survival rate is associated with a treatment-related mortality exceeding >10% and thus leading to a median survival of only 2–8 months.1, 2, 3, 4, 5, 6 Thus, only very few patients with r/r ALL can be cured by standard therapy. To improve patient outcome, new T-cell-based therapies are currently being tested in patients with relapsed/refractory CD19+ B-precursor ALL. Early clinical trials with chimeric antigen receptor-transduced T cells targeting CD19 resulted in a remission rate of 70–90%.7, 8, 9, 10 In addition, the T-cell-engaging bispecific antibody construct blinatumomab can also redirect T cells to recognize and lyse CD19+ B-precursor ALL cells. The first dose finding trial with blinatumomab as monotherapy in r/r ALL patients resulted in a haematological remission rate of 69%11 and a 30-month survival of 22.2%.12 The confirmatory multinational trial demonstrated a clinical activity of blinatumomab in r/r B-ALL patients with a complete remission (CR) of 42% after two cycles of blinatumomab. Response was mainly seen in patients with a bone marrow blast <50% (CR/CR with partial haematologic recovery (CRh) 73%), whereas patients with >50% BM blast had a response of 29%.13 This could be confirmed by the most recent trial comparing blinatumomab with standard of care chemotherapy. This trial shows an improved overall survival of patients receiving blinatumomab (7.9 months) vs patients treated with chemotherapy (4 months).14 Thus, despite being the most effective therapy in r/r ALL patients in this large phase II trial, 52% of all study subjects in the trials did not respond to blinatumomab. No response was observed in the T-cell compartment, thus blinatumomab is responsible for eradicating CD19+ B-precursor ALL. Within the T-cell compartment, regulatory T cells (Tregs) are a key player in modulating an immune response and have an essential role in the maintenance of immune homeostasis and prevention of autoimmunity. Tregs are characterized by the surface markers CD4/CD25high, CD127dim and the intracellular transcription factor FOXP3, with the limitation of CD25 and FOXP3 being upregulated as well as CD127 downregulated after T-cell activation.15 Activated Tregs have a distinct cytokine pattern with interleukin-10 (IL-10) and tumour growth factor-β being secreted in high amounts when compared with T-helper type 1 (Th1) T cells.16 The mode of suppression is mediated by cell–cell contact-dependent modulation of cytokine production and killing of effector T cells.17, 18
In cancer patients, Tregs may have a role in tumour development and immunosuppression by downregulating the effector cells. In murine tumour models and in patients with melanoma, therapy with CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) antibody leads to a decreased number of intratumoral Tregs and a better tumour control.19 Thus, changes in the number of Tregs may have an impact on the efficacy of immunotherapy. In this study, we asked the questions (i) whether elevated Treg numbers influence the outcome of blinatumomab therapy in r/r B-precursor ALL patients, (ii) how does blinatumomab influence Treg expansion and activity and (iii) can the depletion of Tregs restore the functionality of blinatumumab-triggered T-cell function in B-ALL patients.
Materials and methods
Cells
The ALL cell line NALM6 (DSMZ no.: ACC 128) was cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum (Biochrom, Berlin, Germany). These cells were used as target cells in our activation, proliferation and suppression assays. ALL blasts from six B-ALL patients with peripheral blast counts over 90% were isolated by Ficoll and used as primary target cells. Cell lines were tested for mycoplasma contamination.
Human blood donors, cell counts and cell isolation
Blood was obtained from healthy donors and patients after informed consent (ethical approval no.: 214/12). Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Hypaque (Biochrom). CD4+ T cells and Tregs were enriched by Treg Cell Isolation Kit II (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturer's instructions.
Flow cytometric characterization of T cells
For phenotypic analysis of the Tregs, the following anti-human monoclonal antibody were used: CD4/PerCP (BD Biosciences, Heidelberg, Germany); CD25/FITC (BD Biosciences); CD39/APC (eBioscience; San Diego, CA, USA); FOXP3/PE (BD Biosciences); Helios/Horizon (BioLegend, San Diego, CA, USA); CD127/PeCy7 (BD Biosciences). For analysis of surface markers, cells were incubated at 4 °C for 20 min with a monoclonal antibody. For the staining of the intracellular molecules Fixation/Permeabilization buffers (eBioscience) were used. For T-cell subsets, the fresh isolated PBMCs were washed with phosphate-buffered saline (Sigma, München, Germany)/fetal calf serum 10% (Biochrom) at room temperature. For the surface antigens staining to discriminate the T-cell subsets, CD3/PerCp (BD Bioscience), CD4/Amycian (BioLegend, Fell, Germany), CD8/Pacblue (BD Bioscience), CD45RA/FITC (BD Bioscience) and CD45R0/PE (BD Bioscience) antibodies were incubated at 4 °C for 20 min and evaluated by a fluorescence-activated cell sorter (FACS). Activation markers were analysed after 24 h stimulation with the antibody blinatumomab and target cells as described in the topic proliferation assay. Cells were analysed by the BD FACSCANTOII (BD Biosciences, Heidelberg, Germany) and FlowJo Software (Tree Star Inc., Ashland, OR, USA).
CFSE labelling–proliferation–suppression assays
A total of 1 × 105 cells per well of naive CD4+CD25− cells were labelled with 1μm carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen, Darmstadt, Germany) for 10 min at room temperature and were seeded in a 96-well round bottom plate (Corning Incorporated, Corning, NY, USA) and stimulated with 0.5–1 × 105 cells per well NALM6 or ALL blasts and blinatumomab (50 ng/ml) without further supplements. After 3 days, CFSE signal was analysed by flow cytometry. Proliferation was measured with division index analysed by FlowJo Software (Tree Star Inc.). For suppression assays, Tregs were added at day 0 at a TH cell:Treg cell ratio of 2:1. Suppression was measured 72 h later with the division index method previously described by McMurchy and Megan:20 100−(division index of Th cells cocultured with Tregs/division index of Th cells without Tregs)*100.
Proliferation assay with patient PBMCs: Treg cell depletion
Patient samples were collected on day 0 before the start of blinatumomab therapy. PBMCs were isolated by Ficoll as described previously, and were frozen for later analysis. After thawing frozen PBMCs, all cells were labelled with CFSE. Cells were divided in two groups. As a control, group one was cocultured with NALM6 target cells and blinatumomab at an effector-to-target ratio of 2:1. The second group underwent a Treg cell depletion as described below (depleted Tregs). Cells were stained and analysed by FACS before and after Treg cell depletion.
T-cell depletion
For selection of CD39+ Tregs, the CD39 APC antibody (eBioscience; San Diego, CA, USA) was titrated to bind only to CD39high-positive cells. Anti-APC beads were also titrated to select highly positive CD39-bound Tregs. Bead isolation was carried out according to the manufacturer's instructions. A proliferation assay was performed as described above. Proliferation was also calculated using the division index method.
Transwell suppression assay
CD4 Tregs and non-Tregs from healthy donors were isolated as described above in the section 'Human blood donors, cell counts and cell isolation'. CD4 non-Tregs were CFSE labelled. A total of 0.5 × 105 Tregs were added to the top chamber of a Corning HTS Transwell-96 Tissue Culture Systems (Corning Incorporated) (0.4 μm pore size) along with 0.5 × 105 NALM6 target cells. The control top chamber was loaded with CD4 non-Tregs along with the same amount of NALM6 cells. At the bottom chamber CFSE-labelled CD4 non-Tregs (0.5 × 105) and NALM6 (0.5 × 105) cells were added. Blinatumomab (150 ml) was added to the sera at a concentration of 50 ng/ml. Proliferation of CD4 non-Tregs in the bottom chamber was measured on day 3.
Flow cytometry-based cytotoxicity assay
A total of 2 × 105 CD3 or CD8 effector cells were seeded in a 96-well round bottom plate and stimulated with 1 × 105 CFSE- labelled NALM6 (described above) and 50ng/ml blinatumomab. Tregs were added in concentrations of 1 × 105, 0.5 × 105 or 0.1 × 105, respectively. No Tregs were added for controls. After 72 h, 1 × 105 NALM6-labelled cells were added repeatedly. After 24, 48, 72 and 96 h of stimulation, apoptosis was determined by staining with Annexin V-Alexa647 (BioLegend). Apoptotic target cells were defined as Annexin V+, CFSE+. Staining was measured on a FACS-Canto II and was analysed with the FlowJo Software. Specific reduction of apoptosis was calculated as follows: 100 × ((% Apoptotic target cells in positive control)–(% Apoptotic target cells in Treg samples))/(% Apoptotic target cells in positive control).
Ki67 immunhistochemistry staining
Ki67 immunohistochemistry in bone marrow infiltrates was performed according to standard protocols.
Cytokine staining with cytometric bead array technology
Interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α) and IL-10 production by blinatumomab-redirected T cells was determined from culture supernatants of the proliferation assay, as described previously, after 24 h using Cytometric Bead Array (BD Biosciences) according to the manufacturer's instructions. Quantification of cytokine production was analysed using the FCAP Array v.2.0 Software (SoftFlow, Pécs, Hungary).
Statistics
Statistical analysis
Student's t-tests were used for statistical analysis of in vitro data to determine the statistical significance (P<0.05). Data were analysed by GraphPad Prism 4.03 program (GraphPad Software, San Diego, CA, USA).
All analyses were performed using the statistical software framework R (version 3.0.1). For categorical variables Fisher’s exact test has been used,21 whereas continuous covariates have been tested with the nonparametric Wilcoxon's rank-sum test as implemented in R. A decision tree approach22 was applied to obtain a first overview of the covariate structure in relation to the classification of the responder and non-responder groups using the rpart R package version 4.1-3. The Gini coefficient was used as a splitting criterion. Logistic regression models were fitted using the glm function as implemented in the R base package. Model covariates have been selected by a step-down approach starting with the full model of all covariates significant in the univariate tests. In each step the LRT (likelihood ratio test) has been used to assess marginal significance, and nonsignificant (significance threshold P<0.05) covariates have been dropped iteratively. To obtain an estimate of the expected classification error, a cross-validation approach was applied as implemented in the CVbinary function of the DAAG package. The data were randomly divided into n=10 subsets, where each subset was sequentially removed, whereas the model was refitted to the remaining data and used to predict the response of the omitted patients.
Study approval
Patients
Forty-two patients in the MT103-206, 211, 311 trials (EUDRACT 2009-015989-62, 2011-002257-61, 2013-000536-10) and patients outside the trials were immunomonitored in Würzburg.
Ethics statement
Human studies were performed after written informed consent of the study participants, in accordance with the Declaration of Helsinki, and were approved by the institutional review board of the University hospital Würzburg (No. 214/12).
Results
Between December 2010 and February 2016, 42 patients in our center mainly within and outside the scope of the AMG206, AMG211 and the AMG311 trials were included in these analyses. A comparison of the demographics of our 42 patients with both the 189 patients from the AMG211 (ref. 13) trial and 36 patients from the AMG206 trial (ref. 11) was performed. Table 1 shows the patient baseline characteristics. Gender and age in our patient population were comparable to both named trials. Thirty-six per cent of our patients, 34% of AMG211 trial patients and 42% of AMG206 trial patients had a relapse after prior allogeneic hematopoietic stem cell transplantation. Twelve per cent of the 42 patients had no previous salvage therapy when compared with the 20% in the AMG211 trial and 50% in the AMG206 trial. More than one prior salvage therapy lines were found in 88% of our patients, 80% of the AMG211 trial patients and the 50% of the AMG206 patients. Of the 42 treated patients at our center, 22 (52%) had a CR or CRh within the first two treatment cycles. Among the patients, 81 (43%) in the AMG211 trial and 25 (69%) in the AMG206 trial reached a CR or CRh. A bone marrow blast count of >50% was documented in 26 (62%) of our patients. This is similar to the AMG211 population with 130 (69%) patients out of 189. In the AMG206 trial, the median blast count was 77%. The blast count of > or <50% was not reported in the AMG206 trial.
Increased percentage of Tregs in peripheral blood is associated with a reduced response of relapsed ALL patients to blinatumomab
Blood samples were collected from 42 r/r B-precursor ALL patients within the blinatumomab MT103-206 and MT103-211 trials, and later on were collected before blinatumomab therapy. Samples were analysed by an immunomonitoring panel as described in the Material and methods section. The Treg cell detection and gating strategy is shown in Figure 1. In analysing the lymphocyte subsets there was no correlation between the absolute number of T cells and a response to blinatumomab (P-value 0.07; Table 2). In addition, no correlation was seen for CD4+ T-cell (P-value 0.42) and CD8+ T-cell subsets (P-value 0.6), and also with these compartments of both naive and effector T cells marked by CD45RA/RO expression (naive T cells P-value 0.69; effector T cells P-value 0.68) (Supplementary Figure 1). The proliferation marker Ki67, which is postulated to be a marker for therapy failure in ALL induction therapy,23 could be assessed by immunohistochemistry in bone marrow infiltrates in 26 of the 42 ALL patients. There was again no association between the Ki67 values and response to blinatumomab (P-value 0.92; Supplementary Figure 1). We identified the percentage of Tregs in the peripheral blood (TregPercentageCD25FOXP3) with CD4, CD25 and FOXP3 as the most significant marker to identify and differentiate the response to blinatumomab (P-value 0.0001). The level of significance of CD4 and FOXP3 (TregPercentage) as Treg markers to differentiate and correctly predict the two classes of patients is lower (P-value 0.00024), as shown in Table 2.

Tregs in patient material before therapy. FACS analysis of patient material before therapy. Dot plots show the percentage of CD4+CD25+FOXP3+ Tregs. One exemplary patient from the responder and one of the non-responder group is shown.
To evaluate whether known clinical factors might influence the response in r/r ALL patients to salvage therapy,24 we first performed univariate analyses of each factor and then estimated the effect of the covariates in a multivariate regression model.
First, all covariates have been tested separately for significant differences between the responder and non-responder group. Out of nine tested covariates (age, LDH, bone marrow blast infiltration, CD3 absolute numbers, TregPercentageFOXP3, TregPercentageCD25/FOXP3, Tregs/CD3 ratio, sex, allogeneic stem cell transplantation prior therapy), five showed significant differences (Table 2). LDH in particular showed a highly significant increase in the non-responder group (median 402.5 UI/l) compared with the responder group (median 211.5 UI/l). Similarly, a stark increase in TregPercentageFOXP3 could be observed in the non-responder group (median 14.25%) compared with the responder group (median 8.75%). Furthermore, a highly skewed distribution between the genders of the response groups could be observed. In the bigger cohort of the MT211 study, gender was not a predictable marker for responders or non-responders.13
All variables that showed significant differences between the responder and non-responder group have later been included into the logistic regression analysis. To obtain a primary overview of the covariate structure regarding their potential to separate the responder and non-responder group, we first fitted a classification tree including all covariates (regardless of the significance in the univariate tests). The resulting decision tree selected only two variables (TregPercentageCD25/FOXP3 and LDH) where the primary split was defined by the Tregs predicting non-responders on a level of 8.525% or higher with an accuracy of 100% (14/0). The second split turned out to be LDH, which further subclassified responders with LDH<318.5 with 94.7% accuracy. The remaining group of nine patients with a low Treg and high LDH was classified as non-responders with only a low accuracy of 55.6%. In total, the decision tree approach shows an internal accuracy of 88.1%. Cross-validation, however, suggests a pruning of the tree at the first level, leaving only the Tregs as a model covariate. This classifier shows an internal accuracy of 85.7% and an estimated cross-validation accuracy of 81.2%. Nevertheless, the covariate ranking of this rough exploratory analysis is consistent with the results of the univariate tests, where TregPercentageCD25FOXP3 and LDH exhibited the most significant median differences between the groups (Table 2).
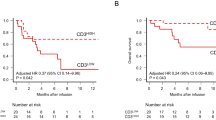
Subsequently, we performed a classical logistic regression analysis to estimate the contribution of the covariates to the risk of response failure. The full model (including all five significant covariates) detected a borderline significant risk association only for TregPercentageCD25FOXP3, whereas gender, LDH Treg/CD3 and bone marrow blast infiltration remained insignificant. The model selection procedure (see Materials and methods section) iteratively dropped nonsignificant covariates and yielded a resulting model with only TregPercentageCD25FOXP3 as a highly significant predictor. This factor is associated with a decrease in therapy responsiveness (OR 0.548 TregPercentage, P-value 0.002). To estimate the expected prediction error, we used a cross-validation approach (see Materials and methods secton). This did yield a cross-validation estimate for the classification accuracy of 81.0% as compared with an internal estimate of classification accuracy of 83.3% (Figure 2).

Classification performance of the final regression model with Tregs as a predictor. (a) Density of the predicted probability for responders (red) and non-responders (blue); dots denote the prediction values of individual patients. (b) Percentage of Tregs (logarithmic scale) in both groups coloured by model-based response prediction with a classification threshold of 50% prediction probability. This yields an internal accuracy of 83.3% and an estimated cross-validation accuracy estimate of 81%.
Tregs are activated by CD19+ B-precursor ALL cell lines and primary B-precursor ALL blasts
We identified that the Treg number can predict the response to blinatumomab in r/r ALL patients. We aimed to understand the effects blinatumomab has on Tregs after being cocultured with primary B-precursor ALL blasts in vitro. CD19 expression was first confirmed on all target cells (data not shown). In the first set of experiments CD4+CD127dimCD25+ T cells from six healthy donors were cocultured with the ALL NALM6 cell line and primary B-precursor ALL blasts from six different patients in the presence or absence of blinatumomab. After 24 h, the expression of CD25, CD69 and PD-1 levels were assessed by flow cytometry. All markers were upregulated in the presence of blinatumomab in the coculture when compared with the isotype control. In detail, PD-1 was upregulated on an average 1.7-fold in Tregs, CD69 expression by 9.6-fold and CD25 expression by 4.5-fold after incubation with primary ALL samples. Upregulation was seen in all tested Treg samples, regardless of whether the ALL cell line NALM6 or primary ALL samples were used (Figures 3 a and b).

Activation and cytokine production of Tregs engaged by blinatumomab. (a) FACS analysis of upregulation of CD69, CD25 and PD-1 on Tregs of a healthy donor cocultured with NALM6 (ALL cell line) cells (red) or ALL primary blasts (blue) after 24 h stimulation with or without blinatumomab. One representative experiment out of seven (b) x-fold MFI of the markers CD69, CD25 and PD-1 of Tregs engaged by blinatumomab NALM6 or ALL cells. No significant difference between stimulation via NALM6 or primary ALL blasts, P-value>0.05. (c) Results of seven different healthy donors and six different ALL blasts from ALL patients. Cytokine profile of TH-1 (CD4+CD127high, CD25low—black) and Tregs (white) after 24 h stimulation with (+) or without (−) antibody and the cell line NALM6. Cytokine production of five different donors measured in the supernatant with cytometric bead array (CBA) technology.
To address the question whether the observed upregulation of Treg activation markers is dependent on the HLA-DR, ICOS-L, B7-H1, CD80, CD86 and CD40 expression (Supplementary Figure 1) on the target cells, both the NALM6 cell lines and all six primary B-precursor ALL samples were stained for these markers. Interestingly, despite variability in expression patterns (Supplementary Figure 1), all tested targets of B-ALL blasts and NALM6 induced Treg activation to the same degree (data not shown). These data show that Tregs are activated by CD19+-targeted cells (antibody blinatumomab present) independent of the expression of HLA-DR, ICOS-L, B7-H1, CD80, CD86 and CD40.
Blinatumomab-activated Tregs secrete high levels of IL-10 but low amounts of TH-1 cytokines
Tregs demonstrate a distinctive cytokine pattern upon activation. Therefore, CD4+CD127dimCD25+ cells and Th cells (CD4+CD25−) from five healthy donors were cocultured with NALM6 and blinatumomab. The culture supernatant was collected after 24 h and analysed for IFNγ, TNF-α and IL-10 by the cytometric bead array assay (Figure 3c) in cocultures supplemented with blinatumomab; Th cells produced the TH-1 cytokines IFNγ on an average of 336 pg/μl (s.e.m. 107), TNF-α on an average of 1050 pg/μl (s.e.m. 331) but only 179 pg/μl of IL-10 (s.e.m. 37.7). In contrast, coculture with purified Tregs supplemented with blinatumomab showed that IL-10 was predominately found with a medium concentration of 270.3 pg/μl (s.e.m. 74.6), whereas IFNγ and TNF-α could be detected in a concentration of 3.9 pg/μl (s.e.m. 101) and 119.5 pg/μl (s.e.m. 48). In a control coculture of both Th- and Treg-purified T cells without blinatumomab, no cytokines were detected in the supernatant.
Blinatumomab-activated Tregs are potent suppressors of autologous CD4CD25− cells
The hallmark of Tregs is the suppression of conventional T cells (TH cells) via contact inhibition and/or cytokines. Therefore, 1 × 105 isolated CFSE-labelled Th cells from seven healthy donors were cocultured with CD19+ target cells NALM6 and ALL blasts from seven ALL patients. Resting isolated Tregs were added to the CFSE-labelled effector T cells at a ratio of 2:1 (Th cell:Treg cell) at the initiation of the coculture. After 72 h, CFSE proliferation was measured (Figure 4) and a suppression capacity was calculated as described. In all cultures, regardless of using the NALM6 ALL cell line or primary B-ALL as targets, blinatumomab-activated Tregs exerted a significant suppression of Th proliferation (53.6% NALM6, s.e.m. 6.5 and 55.1% ALL, s.e.m. 6.5; n=7). Titration of Tregs with a range of 2:1–1:16 into Th1 cultures in the presence of blinatumomab demonstrated a dose-dependent decrease of Treg suppression activity from 40.2 to 7.54% (Figure 4d). These findings indicate that blinatumomab-activated Tregs are able to suppress Th1 function in a dose-dependent manner. Similar suppression results were seen with bulk CD3 T cells and CD8 T cells (data not shown).

Suppression of proliferation in TH-1 cells cocultured with Tregs. (a) CFSE proliferation assay of TH-1 cells cocultured with ALL cells with blinatumomab (black) or without blinatumomab (grey) after 3 days. (b) Antibody-stimulated TH-1 cells (black) cocultured with ALL cells and Tregs and TH-1 cells unstimulated without antibody cocultured with ALL and Tregs as a control. (a and b) One representative experiment out of six. (c) Suppression of proliferation in CD4CD25− cells in six healthy donors cocultured with blinatumomab-redirected Tregs, primary ALL blasts (blue) and NALM6 (red), N=6. (d) CFSE proliferation of TH-1 cells suppressed by titration of Tregs in different ratios to Th cells. Different experiments of five healthy donors.
Tregs suppression does not use soluble factors
To evaluate whether Tregs activated by blinatumomab are able to suppress conventional T cells in a contact-dependent or contact-independent manner, we performed a transwell experiment. The bottom chamber was loaded with freshly isolated and CFSE-labelled conventional T cells activated by blinatumomab. At the top chamber, Tregs or Th1 cells were cultured in media with blinatumomab, and the NALM6 target cells as a control. After 72 h, Th cells in the bottom chamber were collected and proliferation was analysed by FACS. There was no influence on the proliferation capacity of the conventional T cells whether it was incubated with Tregs or Th1 cells at the top chamber with a division index of 0.667 and 0.6198 (Supplementary Figure 2).
Overall, these findings indicate that Tregs suppress Th cells after activation by blinatumomab-targeted ALL cells in a contact-dependent manner and therefore might have a major role for the reduced efficiency of blinatumomab therapy in patients with elevated Treg frequencies.
Tregs activated with blinatumomab reduce the cytotoxic activity of effector T cells
Blinatumomab-induced lysis of B-ALL is mediated by cytotoxic T cells. Therefore, we developed an assay in which Tregs were activated by blinatumomab for 72 h in the presence of target cells before being placed together with fresh NALM6 target cells. Activated Tregs derived from four healthy donors were able to reduce significantly the lytic activity of both purified CD3 and to a greater extent also CD8 T cells (Figure 5). In contrast, non-activated Tregs failed to reduce the blinatumomab lytic activity of T cells (data not shown).

Tregs activated with blinatumomab reduce the cytotoxic activity of effector T cells. (a) Untouched isolated CD3 and CD8 cells were cocultured with the CFSE-labelled target cell line NALM6. Tregs were added at a 1:2 Treg-to-effector cell ratio and activated with 50 ng/ml blinatumomab. After 3 days, Treg preincubation samples were loaded with CFSE-labelled NALM6 and apoptosis was measured after 24 h by Annexin V staining (n=4). (b) Significantly reduced apoptosis was detected in samples coincubated with Tregs, CD3 or CD8 cells and blinatumomab.
Depletion of Tregs in patient samples increased proliferation of PBMCs after blinatumomab
In the clinical studies, patients with elevated levels of Tregs had an inferior outcome of treatment by blinatumomab. Therefore, we addressed the question if depletion of Tregs would be a feasible strategy to overcome this limitation. To this end, we developed a strategy to deplete Tregs in samples of patients who failed blinatumomab therapy and showed elevated levels of Tregs in the peripheral blood. At first, we focussed on the activation marker CD25. We excluded this possibility as it was technically not sufficient to deplete Tregs significantly in frozen samples. Therefore, we used CD39 (used exclusively on Tregs) to develop a depletion strategy by using the magnetic bead separation technique with frozen patient samples, which was drawn before blinatumomab therapy (Figure 6a). About 50% Treg depletion was achieved in patient samples after depletion of CD39+ cells, reducing the percentage of Tregs from 15.7% (s.d. 2) to 7.3% (s.d. 1.1), which resembles the physiological percentage of Tregs. Next, we compared the proliferative response of CD3+ T cells from non-responding patient PBMCs before (untouched PBMCs) and after depletion of CD39+ Tregs (Treg-depleted PBMCs). In total, three patient samples could be analysed with high amounts of Tregs and no clinical response to blinatumomab therapy. For analysis, the proliferation rate of untouched PBMCs was set to 100% proliferation. CFSE dilution was measured after 72 h and analysed with the division index method (see Materials and methods section and Figure 6b).

Tregs suppress proliferation of CD3 cells in patient PBMCs engaged with blinatumomab. (a) Depletion of Tregs in patient PBMCs. CD39 was coexpressed on Tregs in some patient PBMCs. CD39+ cells were depleted out of the PBMCs with CD39 staining and MACS bead technology. Proliferation was measured in CFSE-stained PBMCs with (blue) or without (red) Treg depletion (b) cocultured with NALM6 and +/- blinatumomab. (c) Significantly increased proliferation in CFSE-stained PBMCs after depletion of Tregs in comparison with not depleted PBMCs, P-value<0.05. Proliferation assay of PBMCS from three different ALL patients cocultured with NALM6 cells and the antibody blinatumomab. Proliferation was measured by CFSE dilution after 3 days (described in Materials and methods section). Tregs were depleted by CD39 selection and reincubated with PBMCs in the ratio 1:2 Tregs to PBMCs up to 1:16 Tregs to PBMCs.
Depletion of Tregs in patient samples restored the blinatumomab-triggered proliferation activity of r/r ALL patient T cells in PBMCs. In all tested samples, the proliferation rate of the CD3+ T cells could be increased to 1.37-fold (s.d. 0.102) compared with the proliferation of untouched PBMCs. This could be shown in both CD4 and CD8 cells (data not shown). Conversely, titration of positively selected CD39 Tregs to PBMCs resulted in a dose-dependent suppression. In the presence of high levels of Tregs (2:1; PBMC Treg depleted:Treg), proliferation decreased with a baseline of 27.7%. The proliferation of PBMC Treg depleted could be restored with a proliferation of 81.7% (s.d. 7.4) when the Tregs were diluted to a ratio of 16:1 (Figure 5c). Thus, Tregs in patients were functional, could be redirected to blinatumomab and suppress proliferation of PBMCs.
Discussion
The explorative phase II trial AMG103-206 and the confirmatory phase II trial AMG103-211 demonstrated for the first time that monotherapy with a bispecific T-cell-engaging antibody results in clinically relevant tumour response and CRs in over 40% of r/r ALL patients. Results show that ~50% of all treated patients respond to blinatumomab, resulting in a favourable long-term outcome in this dismal patient population. These data have now also been confirmed in a larger phase III trial in r/r ALL by demonstrating that blinatumomab results in a survival benefit when compared with standard chemotherapy-treated patients. In our study, we could show that a single-cell population identified by enumeration of CD4CD25 and FOXP3 Tregs in the peripheral blood is predictive for a response to blinatumomab therapy. Furthermore, a cutoff of 8.525% can be used to identify all blinatumomab responders and simultaneously identify 70% of patients who do not benefit from blinatumomab therapy. Furthermore, we could also show that Tregs suppress the blinatumomab-induced leukaemia control in vitro and that depletion of Tregs in blinatumomab-non-responding ALL patients restored the T-cell proliferation. Flow cytometry analyses of patient PBMCs before and during treatment, to describe the T-cell compartment, were previously performed within blinatumomab trials, starting with the minimal residual disease (MRD) setting. The authors did not find any difference between responders and non-responders in the absolute counts of T cells before the start of blinatumomab25 and also in all T-cell subsets tested. We also did not find any correlation of responder patients to the initial T-cell numbers and different T-cell subsets such as CD4, CD8, naive and memory T cells (Supplementary Figure 2). Blinatumomab as a T-cell engager increased the absolute counts of CD3 cells and the percentage of activated T cells in peripheral blood in the MRD setting during the first cycle.25 Mostly, T effector memory cells CD45RA−/CD197− could be detected as the expanding CD8 population. Zugmaier et al.12 found a positive correlation between the expanding capacity of CD3 T cells to blinatumomab and the long-term surviving r/r ALL patients, but failed to identify any predicting T-cell markers before therapy for the non-responding MRD-positive patients. In contrast to these results, we could demonstrate that a high percentage of Tregs in non-responding patients is predictive to determine the outcome of blinatumomab. Based on the observation that Tregs may have an influence to blinatumomab therapy, we verified whether Tregs can be activated with this bispecific antibody. Activation markers such as CD25 and CD69 were upregulated in response to ALL blasts coated with blinatumomab, resulting in marked suppression of proliferation of effector T cells. In addition, there is no difference whether Tregs were activated either with different primary ALL blasts with different expression levels of costimulatory/inhibitory ligands or ALL cell lines. Thus, the identified mechanism is not due to different ALL blasts and their variety in costimulatory ligands but rather due to absolute numbers of Tregs in the given individual patient. In both the MRD setting and in the r/r ALL setting, blinatumomab responders demonstrated a marked T-cell proliferation, whereas the non-responders failed to show a significant increase in absolute T-cell numbers upon blinatumomab therapy. This observation has been documented both in the MRD setting25 and in the r/r setting in patients who achieved either a molecular remission or a CR/CRh+ in the respective setting. In our study, the high amount of Tregs dampens the proliferative capacity of T cells in response to blinatumomab. We could demonstrate in vitro that high amounts of Tregs reduce the proliferation of patient-derived T cells. Thus, it is conceivable that low proliferative response invertible correlates with the outcome of blinatumomab therapy. We also could generate in vitro data that show a significantly reduced lysis capacity of CD3 and CD8 effector T cells if preactived Tregs were present in the vials. These data suggest a second mechanism of blinatumomab treatment failure by Tregs (Figure 5). In our study, we screened for other predictive markers of therapeutic success just as part from the T-cell compartment. To this end, as described previously, a higher tumour burden was seen more frequently in r/r ALL not responding to blinatumomab13 and could be confirmed in our analysis (Table 2). Interestingly, high Ki67 expression as a marker for proliferation of tumour cells in the bone marrow did not correlate with the response to blinatumomab (Supplementary Table 3). This marker has been shown to predict response to treatment of naive B-CLL patients with an advanced stage and in line with a poor prognosis due to failed therapies.26
Mechanisms of immunosuppression by Tregs are the secretion of inhibitory cytokines, the induction of cytolysis, metabolic disruption and targeting dendritic cells.27 The cytokine profile of the Tregs redirected with blinatumomab in coculture with NALM6 showed the secretion of IL-10, the hallmark cytokine of Tregs. IL-10 has shown to mediate Treg-induced T-cell suppression but other reports have shown that IL-10 can also restore T-cell immunity.28 The TH-1 cytokines IFN-γ and TNF-α were rarely produced by Tregs in contrast to CD4/25− cells. The results are in concordance with a study in which Tregs redirected with a CD3xPSCA bispecific antibody showed the same cytokine profile as in our study.29 IL-10 production is not the only factor in mediating blinatumomab-induced suppression, as our transwell experiments showed that cell-to-cell contact-mediated suppression is essential for suppression. Whether the granzyme B-mediated kill function of Tregs27, 30 as a cell-to-cell contact mechanism has a major role in inducing the suppression remains unclear.
At our centre, 67% of the patients treated within the blinatumomab trials had low Treg numbers (defined with a cutoff of 8.525%), and among those with low Treg numbers, the response rate was 78.6%. This very high response rate within this subgroup of r/r ALL patients has also been reported for r/r ALL patients treated with chimeric antigen receptor (CAR) T-cell therapy.7, 8, 9, 31 Nevertheless, patients with high Treg numbers, using the same cutoff of 8.525% Tregs in the peripheral blood had a 100% failure rate to blinatumomab. Thus, why would CAR-T-cell therapy overcome this potential resistance mechanism of redirected T-cell therapy? At first, all CAR-T trials use a preparation chemotherapy backbone, which always includes cyclophosphamide and fludarabine. Both chemotherapy agents have been shown to reduce Treg numbers32, 33, 34 in solid cancer and CLL patients. Furthermore, a major difference between both successful approaches using T cells to control leukaemia is that CAR-T cells are cultured for several weeks in an environment with CD3/CD28 beads, which favours the expansion of functional T cells and reduces Treg population. Nevertheless, the current CAR-T-cell reports in r/r adult patients do neither reveal any detailed information on the T-cell subset infused to patients nor correlate response to the T-cell phenotype of the infused CAR-T product.
The development of a biomarker test to predict the outcome of blinatumomab therapy in r/r ALL has to be evaluated prospectively. Should these trials confirm our key study result for predicting the response to blinatumomab this could (i) identify upfront patients who will benefit from blinatumomab, (ii) identify patients who would benefit from other approaches in r/r ALL such as inotuzumab35 and CAR-T-cell therapy and (iii) would be cost effective.
In addition, if such prospective trials can confirm the biomarker approach, these would also suggest that blunting the Treg function in vivo might be a potential strategy to convert patients with a low chance of responding to high responders with blinatumomab. One opportunity is a treatment of patients with cyclophosphamide or fludarabine before blinatumomab therapy to reduce Tregs.33, 34 In addition, checkpoint inhibitors, such as PD-1-blocking antibodies, may also prevent immunosuppressive effects of the blinatumomab-activated Tregs. Furthermore, PD-1 blockade has also been shown to induce a T-cell response to neoantigens in melanoma and non-small-cell lung cancer patients.35, 36 Although ALL shows an average low rate of neoantigens when compared with melanoma and non-small-cell lung cancer,37 the combination therapy of T-cell-engaging bispecific antibody construct with PD-1 may still induce long-lasting immune surveillance in this hard-to-treat population of r/r leukaemia.
In summary, we can show that Tregs have a crucial role both on a cellular level as well as in the clinical setting in ALL patient treatment with blinatumomab. The percentage of Tregs that predicts responders before blinatumomab therapy could become an important biomarker to select patients (i) for treatment with a higher chance to respond to the costly bispecific antibodies and also (ii) not expose patients with a small chance to respond but to develop significant toxicity. A larger clinical study is necessary to further validate this promising marker for clinical treatment with blinatumomab in ALL patients.38
References
Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, Durrant IJ et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 2007; 109: 944–950.
Gökbuget N, Stanze D, Beck J, Diedrich H, Horst HA, Hüttmann A et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood 2012; 120: 2032–2041.
Kantarjian HM, Thomas D, Ravandi F, Faderl S, Jabbour E, Garcia-Manero G et al. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer 2010; 116: 5568–5574.
O'Brien S, Thomas D, Ravandi F, Faderl S, Cortes J, Borthakur G et al. Outcome of adults with acute lymphocytic leukemia after second salvage therapy. Cancer 2008; 113: 3186–3191.
Oriol A, Vives S, Hernandez-Rivas JM, Tormo M, Heras I, Rivas C, Tormo M, Heras I, Rivas C et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica 2010; 95: 589–596.
Tavernier E, Boiron JM, Huguet F, Bradstock K, Vey N, Kovacsovics T et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia 2007; 21: 1907–1914.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371: 1507–1517.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3: 95ra73.
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118: 4817–4828.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385: 517–528.
Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol 2014; 32: 4134–4140.
Zugmaier G, Gökbuget N, Klinger M, Viardot A, Stelljes M, Neumann S et al. Long-term survival and T-Cell kinetics in adult patients with relapsed/refractory B-precursor acute lymphoblastic leukemia who achieved minimal residual disease response following treatment with Anti-CD19 BiTE® antibody construct blinatumomab. Blood 2015; 126: 2578–2584.
Topp MS, Gökbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 2015; 16: 57–66.
Topp MS, Stein A et al. Blinatumomab improved overall survival in patients with relapsed or refractory Philadelphia negative B-cell precursor acute lmyphoblastic leukemia in a randomized open label phase 3 study (Tower). EHA 2016; abstract S149.
Miyara M, Sakaguchi S . Natural regulatory T cells: mechanisms of suppression. Trends Mol Med. 2007; 13: 108–116.
Annacker O, Pimenta-Araujo R, Burlen-Defranoux O, Barbosa TC, Cumano A, Bandeira A . CD4+ CD25+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J Immunol 166: 3008–3018.
Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H et al. TGF-β 1 plays an important role in the mechanism of CD4+ CD25+ regulatory T cell activity in both humans and mice. J Immunol 172: 834–842.
Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ . Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21: 589–601.
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 2013; 210: 1695–1710.
McMurchy AN, Megan KL . Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol 2012; 42: 27–34.
Fisher RA . The logic of inductive inference. J R Stat Soc Ser A 1935; 98: 39–54.
Breiman L, Friedman JH, Olshen RA, Stone CJ Classification and Regression Trees. Wadsworth: Belmont, CA, USA.
Nowicki M, Miśkowiak B, Kaczmarek-Kanold M . Correlation between early treatment failure and Ki67 antigen expression in blast cells of children with acute lymphoblastic leukaemia before commencing treatment. A retrospective study. Oncology 2002; 62: 55–59.
Gökbuget N, Stanze D, Beck J, Diedrich H, Horst HA, Hüttmann A et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood 2012; 6 120: 2032–2041.
Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012; 119: 6226–6233.
Niewiadomska H, Mirowski M, Kulczycka D, Najder M, Balcerczak E, Błoński JZ . Some oncogene and tumour suppressor gene protein products expression in B-cell chronic lymphocytic leukaemia. Cytobios 2000; 103: 159–168.
Vignali DA, Collison LW, Workman CJ . How regulatory T cells work. Nat Rev Immunol 2008; 8: 523–532.
Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S et al. IL-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell 2011; 20: 781–796.
Koristka S, Cartellieri M, Theil A, Feldmann A, Arndt C, Stamova S et al. Retargeting of human regulatory T cells by single-chain bispecific antibodies. J Immunol 2012; 188: 1551–1558.
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007; 27: 635–646.
Maude SL, Teachey DT, Porter DL, Grupp SA . CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015; 125: 4017–4023.
Madondo MT, Quinn M, Plebanski M . Low dose cyclophosphamide: mechanisms of T cell modulation. Cancer Treat Rev 2016; 42: 3–9.
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 2007; 56: 641–648.
Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005; 106: 2018–2025.
Kantarjian H, Thomas D, Jorgensen J, Kebriaei P, Jabbour E, Rytting M et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic. leukemia 2013; 119: 2728–2736.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369: 134–144.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV et al. Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015; 373: 1627–1639.
Acknowledgements
We thank the patients and their families for the participation. JD was supported by Else Kröner-Fresenius-Stiftung/Else Kröner Forschungskolleg Würzburg and by the Interdisciplinary Center for Clinical Research Würzburg. The support by DFG (TR124/B2 to MD, TD and TR124/A4 to MST) is gratefully acknowledged.
Author contributions
Conceived and designed the experiments: JD, TB and MST. Performed the experiments: JD, TB, AR, EH and FE. Analysed the data: JD, TB, TM MD, TD and MST. Contributed reagents/materials/analysis tools: HE and MST. Treated patients: JD, LR, HE and MST. Wrote the paper: JD, MD, TM, TD and MST. All authors have read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MST is an advisory board member, an honorary speaker and a travel support at Amgen. JD, TB, TM, MD, TD, HE, FE, FE, EH and AR declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Duell, J., Dittrich, M., Bedke, T. et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 31, 2181–2190 (2017). https://doi.org/10.1038/leu.2017.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.41
This article is cited by
-
Bi- and Tri-specific antibodies in non-Hodgkin lymphoma: current data and perspectives
Blood Cancer Journal (2024)
-
Real-world analysis of teclistamab in 123 RRMM patients from Germany
Leukemia (2024)
-
New immune cell engagers for cancer immunotherapy
Nature Reviews Immunology (2024)
-
One-Step Immunoassay for the Detection of SARS-CoV-2 Nucleocapsid Protein Using Screened Fv-Antibodies
BioChip Journal (2024)
-
Emerging therapies targeting the delta-like ligand 3 (DLL3) in small cell lung cancer
Journal of Hematology & Oncology (2023)
