
Abstract
Prediction of responses to chimeric antigen receptor (CAR)-T cell therapies is essential to maximize their therapeutic efficacy for diffuse large B-cell lymphoma (DLBCL). While several tumor-intrinsic risk factors of resistance and/or early relapse have been identified, clinically useful markers that determine potential activity of CAR-T cells have not been fully investigated. T-cell property at the time of leukapheresis may serve as such a marker. Therefore, we evaluated the clinical impact of CD3+ cell count in peripheral blood at leukapheresis on clinical outcomes of CAR-T cell therapy. In total, 44 patients with relapsed or refractory (r/r) DLBCL who received tisagenlecleucel at Kyoto University Hospital were included. According to CD3+ cell counts, patients were categorized into CD3LOW and CD3HIGH groups with a threshold of 553/μL, based on receiver operating characteristic curve analysis. 1-year progression-free survival was significantly higher in the CD3HIGH group than the CD3LOW group (68.3% vs. 17.3%; adjusted hazard ratio [aHR], 0.37; p = 0.042). Overall survival was also superior in the CD3HIGH group (aHR, 0.24; p = 0.043). Moreover, higher CD3+ cell counts at leukapheresis were associated with significantly higher lymphocyte counts in peripheral blood at day 7 after CAR-T cell infusion (median 860 vs. 420/μL, P = 0.021), suggesting more extensive expansion of infused CAR-T cells in vivo. In conclusion, we demonstrated that the CD3+ cell count at leukapheresis predicts both expansion of CAR-T cells after infusion and outcomes of CAR-T cell therapy, and are useful for building comprehensive therapeutic strategies at the time of leukapheresis.
Similar content being viewed by others


Introduction
Diffuse large B cell lymphoma (DLBCL) is curable with standard anthracycline-based chemotherapy regimens, but patients with relapsed or refractory (r/r) DLBCL still have dismal prognosis, even with intensified chemotherapy and autologous or allogeneic stem cell transplantation1. CD19-directed chimeric antigen receptor (CAR)-T cell therapies have remarkably improved prognosis for a subset of patients with r/r DLBCL2. While responders to CAR-T cell therapies have promising prognosis, those with incomplete responses have grim prognosis. Moreover, even in patients who once responded to CD19-directed CAR-T therapies, about 20% are reported to experience relapses later3,4,5. Therefore, a risk-stratified approach, based on early identification of high-risk patients for resistance or relapse to CAR-T cell therapies, are required to optimize them.
Recently, several prognostic clinical factors for CAR-T cell therapy, mostly focused on patient characteristics or disease status, have been reported. For instance, high tumor volume at infusion is associated with decreased efficacy of CAR-T cell therapy, which suggests that disease control before administration of CAR-T cells is important for better responses6,7. Certain genetic alterations, such as TP53 mutations, have been reported as tumor-intrinsic biomarkers that inform inferior responses8. Such parameters on the tumor side have been rapidly established. However, efficacy of CAR-T cell therapy is determined not only by tumor-intrinsic factors, but also by the parameters on the CAR-T cell side and the latter have not been fully analyzed9. Moreover, it is essential to predict efficacy of CAR-T cell therapies as early as possible, i.e., at leukapheresis, and to optimize treatment strategy before and after therapies in order to maximize their effects, because candidates for CAR-T cell therapy often have highly refractory, progressive disease, as well as frequent complications.
Therefore, in this study, we focused on the prognostic value of CD3+ cell count in peripheral blood at leukapheresis on outcomes of CAR-T cell therapy. Our findings provide a novel biomarker that enables us to predict responses as early as the time of leukapheresis. This marker should help to improve outcomes for CAR-T eligible patients with r/r DLBCL.
Patients and methods
Study cohort
This study was performed by analysing all consecutive patients with r/r DLBCL who received tisagenlecleucel at Kyoto University Hospital, Kyoto Japan, between December 2019 and October 2021. Follow-up time was fixed in December 2021. The diagnosis of DLBCL was based on the WHO classification of tumors of hematopoietic and lymphoid tissues (revised 4th edition)10. All patients were relapsed or refractory to two or more prior lines of therapy. This study was approved by the Ethics Committee of Kyoto University, and was conducted in accordance with the principles of the Declaration of Helsinki. Informed consent was obtained from all the patients.
Endpoints and variables
The primary endpoint of this study was progression‐free survival (PFS). Overall survival (OS) was evaluated as a secondary endpoint. PFS was defined as the time from the date of CAR-T infusion to the date of documented disease progression, relapse, death, or the last date of follow‐up. OS was calculated from the date of CAR-T infusion to the last date of follow‐up or death. Disease status at leukapheresis, at infusion, and after infusion was assessed using the Revised Response Criteria for Malignant Lymphoma11. Progression of relapse was defined based on morphological and clinical evidence of disease activity. All variables shown in tables and figures were retrospectively obtained from patient records. To simplify risk stratification, we divided our patients into two risk groups (CD3LOW and CD3HIGH group) by employing the receiver operating characteristic (ROC) curve of CD3+ cell count at leukapheresis for progression-free survival (PFS) at 1 year.
Statistical analysis
Continuous variables were summarized using medians and ranges, and categorical variables were summarized as counts and percentages. For comparisons between groups, patients and disease characteristics were compared using the Mann‐Whitney U test for continuous variables and Fisher’s exact test for categorical variables. Correlation between CD3+ cell count at leukapheresis and lymphocyte count after infusion were analyzed using Pearson’s correlation. Probabilities of PFS and OS were estimated using the Kaplan‐Meier method, and were compared between groups using the log‐rank test. In multivariate analysis, Cox proportional hazards models were used to evaluate the effects of CD3+ cell count in peripheral blood at leukapheresis on PFS and OS in combination with other clinically relevant variables, including prior lines of chemotherapies at leukapheresis (0–3 or > 3) and disease status at leukapheresis (complete response [CR]/partial response [PR] or stable disease [SD]/progressive disease [PD]). Statistical significance was set at p < 0.05. All statistical analyses were performed using R (version 3.1.1; R Development Core Team) and EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan)12.
Results
Patient characteristics
In total, 44 patients were included in this study (Table 1). The study cohort comprised 23 females and 21 males, with a median age of 60 years (range, 20–73 years). The median CD3+ cell count in peripheral blood at leukapheresis was 687/μL (range, 60–2234). According to the ROC curve of CD3+ cell count for PFS at 1 year, the cut off value was set at 553/μL. Then, we defined patients with CD3+ cell counts ≤ 553/μL as the CD3LOW group (20 patients), and those with higher CD3+ cell counts as the CD3HIGH group (24 patients). There was no significant difference between the two groups in age at infusion. In terms of DLBCL characteristics, the proportions of patients with germinal center B-cell-like (GCB) DLBCL, double-expressor lymphoma (DEL), transformed follicular lymphoma (tFL) were comparable between the two groups (P = 0.545, 0.475, and 0.734, respectively). There were no significant differences between groups in prior lines of chemotherapy at leukapheresis, or in disease status at leukapheresis, or infusion. Time from diagnosis to leukapheresis was significantly longer in the CD3HIGH group compared with the CD3LOW group (P = 0.036). Lymphocyte depletion protocol was composed of fludarabine with cyclophosphamide (Flu/Cy, n = 37) and bendamustine-based regimens (n = 5), and was not administered in two patients with exceptionally low lymphocyte counts. The characteristics of CAR-T cell products in each group are shown in Supplementary Table 1. The occurrence or grade of cytokine release syndrome (CRS) and the use of tocilizumab in each group are shown in Supplementary Table 2. We used tocilizumab if patients had continuous high fever refractory to conservative treatment or developed grade 2 CRS.
OS and PFS according to the CD3+ cell count at leukapheresis
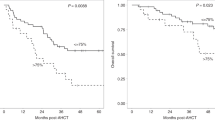
We compared effects of CD3+ cell count at leukapheresis on survival after CAR-T cell infusion, and found that the 1‐year PFS was significantly lower in the CD3LOW group (17.3%, 95%CI 1.50–48.3%) than in the CD3HIGH group (68.3%, 95%CI 44.8–83.5%; p = 0.032) (Fig. 1A). After adjusting prior lines at leukapheresis (0–3 or > 3) and disease status at leukapheresis (CR/PR or SD/PD) as covariates, the adjusted hazard ratio (aHR) of PFS was 0.37 (95% CI: 0.14–0.96, p = 0.042) for the CD3HIGH group compared to the CD3LOW group (Table 2). When CD3+ cell count was treated as a continuous variable, aHR was 0.90 (95% CI: 0.81–1.00, p = 0.052) for every 100/μL increase.

Effects of CD3+ cell counts in peripheral blood at leukapheresis on survival. Kaplan–Meier estimates of progression-free survival (PFS) (A) and overall survival (OS) (B) according to the number of CD3+cells at leukapheresis.
Univariate analysis for OS showed a trend for worse 1‐year OS in the CD3LOW group (54.9%, 95% CI 29.2–74.7%) compared to the CD3HIGH group (84.2%, 95% CI 47.0–96.2%; p = 0.054) (Fig. 1B). Multivariate analysis revealed that lower CD3+ cell counts significantly reduced OS (Table 2). aHR for OS in the CD3HIGH group versus the CD3LOW group was 0.24 (95% CI 0.06–0.95, p = 0.043) (Table 2). When CD3+ cell count was treated as a continuous variable, aHR was 0.88 (95% CI 0.75–1.03, p = 0.102) for every 100/μL increase.
The clinical courses of all patients are summarized in Fig. 2 and detailed disease status at apheresis and CAR-T infusion, and post-apheresis and lymphocyte depletion therapy are shown in Supplementary Table 3. The cause of death was mainly disease relapse, except for 2 cases with myelodysplastic syndrome (No. 2) and severe cytokine release syndrome (No. 44). In the CD3HIGH group, only one patient of 10 in CR/PR (10.0%) at infusion relapsed after CAR-T cell infusion, while four patients of five in SD/PD (80.0%) relapsed after infusion. In the CD3LOW group, the proportions of patients who experienced disease progression after infusion were similar in CR/PR and SD/PD [2 of 4 patients in CR/PR (50.0%); 7 of 16 in SD/PD (43.8%)]. Interestingly, regarding Hans cell-of-origin classification, among 28 patients in SD/PD at infusion, patients with GCB-phenotypes responded more favorably than those with non-GCB-phenotypes [10 of 14 patients (76.9%) vs. 3 of 14 patients (23.1%), P = 0.021]. Such a difference regarding the classification was not observed in patients treated in CR/PR at infusion.

Clinical course of patients according to CD3+ cell counts at leukapheresis. Survival time without disease progression (yellow bars) and survival time after-progression (brown bars) after CAR-T infusion of individual patients are shown. Patients were divided into 2 groups (CD3HIGH and CD3LOW) based on CD3+cell counts at leukapheresis. Green dots indicate CD3+cell counts at leukapheresis of individual patients. Disease status at infusion [complete response (CR)/partial response (PR) or stable disease (SD)/progressive disease (PD)] of each patient is shown at the bottom.
Impact of CD3+ cell count at leukapheresis on lymphocyte expansion after CAR-T cell infusion
Because CD3+ cell count in peripheral blood at leukapheresis may reflect the function of CAR-T cells, we sought to analyze effects of CD3+ cell count at leukapheresis on lymphocyte expansion after infusion of CAR-T cells. Lymphocyte counts in peripheral blood at day 7 were significantly higher in the CD3HIGH group (median 860/μL, range 0–2610/μL) than in the CD3LOW group (median 420/μL, range 0–2210/μL; P = 0.021; Fig. 3A), although lymphocyte count at day 0 and number of infused CAR-T cells were comparable between the two groups. Furthermore, CD3+ cell counts in peripheral blood at leukapheresis showed a significantly positive correlation with increased lymphocyte counts from day 0 to day 7 after infusion (P = 0.010) (Fig. 3B). Other blood count data after CAR-T cell infusion in each group are shown in Supplementary Table 4.

Correlation of lymphocyte counts after CAR-T cell infusion with CD3+ cell counts at leukapheresis and their prognostic significance. Box plot showing lymphocyte count at day 7 after infusion in CD3LOW and CD3HIGH group patients (A). Scatter plot showing the correlation with CD3+ cell counts in peripheral blood at leukapheresis and lymphocyte counts in peripheral blood at day 7 after infusion (B). Kaplan–Meier estimates of OS according to lymphocyte count at day 7 (> 1000/μL or ≤ 1000/μL) (C).
Knowing the relationship between CD3+ cell count at leukapheresis and lymphocyte increase after infusion, the next step was to analyze the impact of lymphocyte count at day 7 on PFS. 1‐year PFS in patients with lymphocyte counts at day 7 > 1000/μL (81.8%, 95% CI 44.7–95.1%) was significantly higher than in patients with day 7 counts ≤ 1000/μL (38.0%, 95% CI 19.1–56.8%, P = 0.040) (Fig. 3C). The superiority of PFS observed in patients with lymphocyte counts at day 7 > 1000/μL remained after adjustment for disease status at the time of CAR-T cell infusion, though not statistically significant (P = 0.094). These results suggest that proliferation of CAR-T cells after infusion predicted by higher CD3+ cell counts at leukapheresis can also be a prognostic marker for outcomes following CAR-T cell therapy.
Discussion
This study evaluated prognostic impact of CD3+ cell counts in peripheral blood at leukapheresis in patients with r/r DLBCL who later received CAR-T cell therapy. There were two major findings regarding CD3+ cell count in peripheral blood at leukapheresis: (1) greater CD3+ cell numbers resulted in better PFS and OS, and (2) higher CD3+ cell counts at leukapheresis were associated with significantly higher lymphocyte counts at day 7, suggesting expansion of CAR-T cells after infusion. Our results indicate that CD3+ cell counts in peripheral blood at leukapheresis are useful for predicting outcomes and for designing risk‐adapted strategies including CAR-T cell therapy. The novelty of this study is that it revealed the contribution of parameters on the CAR-T cell side, independent of tumor-intrinsic factors, to the outcome of CAR-T cell therapy.
The positive association of CD3+ cell count at leukapheresis and efficacy of CAR-T cell therapy may be driven by at least three factors, including quality of T cells, efficacy of the CAR-T cell manufacturing process, and in vivo environment13. First, decreased CD3+ cell count in peripheral blood, namely a “low quantity of T cells”, can be a surrogate marker of “inferior T-cell quality” as a source of CAR-T cells. In patients with r/r DLBCL, quality of T cells can be impaired by various factors, such as prolonged dysfunction of T cells caused by an increasing number of chemotherapy regimens14,15,16. In this study, CD3LOW group patients tended to have a higher number of chemotherapy lines than CD3HIGH group patients, although the difference was not statistically significant (Table 1).
Second, although collection of sufficient number of CD3+ cells can be achieved via leukapheresis by optimizing leukapheresis procedures even in patients with low CD3+ cell counts17, subsequent ex vivo manufacturing process of CAR-T cells, where T cells are activated, transduced with CAR, and then expanded, can be potentially complicated in patients with low CD3+ cell counts at leukapheresis. Actually, in our cohort, transduction efficiency of CAR was significantly lower (P = 0.02), and CAR expression tended to be lower (P = 0.085) in the CD3LOW group, compared to the CD3HIGH group (Supplemental Table 1). Our findings support previous studies suggesting that efficiency of CAR integration and expression level is low if the leukapheresis product contains T cells with poor proliferative potential18.
Third, patients with low CD3+ cell counts at leukapheresis, who often have longer history of chemotherapies and more refractory lymphoma, may potentially have disadvantageous in vivo environments for growth and efficacy of infused CAR-T cells. A high tumor burden is one of the factors that degrade the environment, leading to higher immune dysregulation with increased serum inflammatory cytokines and tumor interferon signaling, which results in poor CAR-T cell expansion19,20.
Thus, CD3+ cell counts at leukapheresis can be a surrogate marker for quality of T cells, efficacy of CAR-T cell production, and in vivo environment. In addition to that, we showed for the first time that CD3+ cell counts can predict the number of lymphocytes at day 7 after infusion, suggesting the degree of CAR-T cell expansion. In previous studies, in vivo expansion of CAR-T cells after infusion was reported as a vital marker for function of CAR-T cells3,21,22,23, and early increases in lymphocyte counts are useful for estimating treatment responses. However, there is no established method for evaluating the ability of T cells to generate a CAR-mediated anti-tumor response and for predicting in vivo CAR-T cell expansion, so our finding that CD3+ cell counts at leukapheresis predict later expansion will be useful to develop treatment strategies, including CAR-T cell therapy.
Based on these results, comprehensive therapeutic strategies are needed during the entire course of CAR-T cell therapy in order to maximize therapeutic effects of CAR-T cells. Since reduced CD3+ cell counts in peripheral blood, which are often caused by repeated, heavy exposure to chemotherapeutic agents, are a marker for degraded quality of the source of CAR-T cells, T cells should be harvested in a way that fully maintains their functions. In Japan, CAR-T therapy is approved for patients refractory to more than two lines of chemotherapy. However, T-cell harvesting are considered before starting intensive salvage chemotherapy, rather than after sufficient washout periods during or after chemotherapy. Especially for a subset of patients who are expected to be at high risk of future relapse or resistance to conventional therapies, it may be beneficial to preemptively harvest and store high-quality T cells before T cells become exhausted, even without stringently meeting the indication for CAR-T cell therapy. T cells from patients early after diagnosis yield greater expansion of CAR-T cells than those from patients with r/r disease24. These strategies can enhance CAR-T cell function independent of tumor-intrinsic factors and can improve overall treatment efficiency.
This study has several limitations. First, this is a single-center retrospective analysis with a small number of patients and short follow-up time due to the novelty of CAR-T cell therapy, although our center performs more CAR-T cell therapies than most institutions in Japan and eligibility for CAR-T cell therapy is fairly consistent. In this study, effects of disease status and prior lines of chemotherapies at leukapheresis were carefully adjusted in the multivariate analysis evaluating the impact of CD3+ cell count at leukapheresis on survival; however, potential effects of factors associated with tumor intrinsic factors on survival should be further studied in a larger cohort, given the longer time from diagnosis to leukapheresis observed in the CD3HIGH group compared with the CD3LOW group, the different effects of CD3+ T cells number on survival according to the cell of origin (data not shown) and the higher response rate observed in patients with GCB-phenotypes compared with non-GCB-phenotypes when disease status was SD or PD at infusion. Second, we did not assess expansion of CAR-T cells themselves or detailed phenotypes of CD3+ cells after infusion, while most circulating lymphocytes in the first few weeks after CAR-T cell infusion comprise CAR-T cells themselves22,23. In addition, large numbers of T cells without CAR were also suggested to be activated after CAR-T infusion25. Therefore, we consider that evaluating CD3+ cells, which include both CAR-T cells and non-CAR-T cells, are reasonable to assess the entire effect of CAR-T therapy. Phenotypic differences among CD3+ cells are important factors that contributes the expansion of CAR-T cells and the effect of CAR-T therapy. We found that CD4/CD8 ratio at apheresis showed a positive correlation with increased lymphocyte count at day 7 especially in the CD3LOW group (data not shown). Furthermore, previous studies demonstrated that CAR T cell products generated from naïve/stem memory T cells, as compared with unselected T cells, shows superior antitumor activity and increase expansion rates26,27. However, because it is difficult to evaluate theses phenotypes in the clinical setting, we used lymphocyte counts in peripheral blood as a simple biomarker reflecting CAR-T cell proliferation28,29. Detailed cytokine or chemokine assays were not performed.
In conclusion, we demonstrated that CD3+ cell counts at leukapheresis predicts expansion of CAR-T cells after infusion and the overall outcomes of CAR-T cell therapy. Optimizing CAR-T cell therapy at the time of leukapheresis, especially for high-risk patients, should be considered to achieve favorable CAR-T cell responses.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Crump, M. et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 130(16), 1800–1808 (2017).
Sermer, D. et al. Outcomes in patients with DLBCL treated with commercial CAR T cells compared with alternate therapies. Blood Adv. 4(19), 4669–4678 (2020).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377(26), 2531–2544 (2017).
Pasquini, M. C. et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 4(21), 5414–5424 (2020).
Schuster, S. J. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380(1), 45–56 (2019).
Dean, E. A. et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 4(14), 3268–3276 (2020).
Vercellino, L. et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 4(22), 5607–5615 (2020).
Shouval, R. et al. Impact of TP53 genomic alterations in large B-cell lymphoma treated with CD19-chimeric antigen receptor T-cell therapy. J. Clin. Oncol. 40(4), 369–381 (2022).
Green, M. R. & Neelapu, S. S. Not so FASt: Tumor cells resisting death drive CAR T-cell dysfunction. Cancer Discov. 10(4), 492–494 (2020).
Swerdlow, S.H.C.E., & Harris, N.L. et al. (Ed.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th edn. (International Agency for Research on Cancer (IARC), 2017).
Cheson, B. D. et al. Revised response criteria for malignant lymphoma. J. Clin. Oncol. 25(5), 579–586 (2007).
Kanda, Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 48(3), 452–458 (2013).
Mehta, P. H. et al. T cell fitness and autologous CAR T cell therapy in haematologic malignancy. Front. Immunol. 12, 780442 (2021).
Cooke, R. E. et al. Conventional treatment for multiple myeloma drives premature aging phenotypes and metabolic dysfunction in T cells. Front. Immunol. 11, 2153 (2020).
Saito, H. et al. Prolonged lymphocytopenia after bendamustine therapy in patients with relapsed or refractory indolent B-cell and mantle cell lymphoma. Blood Cancer J. 5, e362 (2015).
Suen, H. et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 30(8), 1716–1724 (2016).
Jo, T. et al. Clinical experience of leukapheresis for CD19 CAR-T cell therapy. Rinsho Ketsueki 62(3), 163–169 (2021).
Riviere, I., Gallardo, H. F., Hagani, A. B. & Sadelain, M. Retroviral-mediated gene transfer in primary murine and human T-lymphocytes. Mol. Biotechnol. 15(2), 133–142 (2000).
Jain, M. D. et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood 137(19), 2621–2633 (2021).
Strati, P. & Neelapu, S. S. CAR-T failure: Beyond antigen loss and T cells. Blood 137(19), 2567–2568 (2021).
Bansal, R. G. A. et al. Peak absolute lymphocyte count post CAR-T is associated with clinical response and survival outcome in aggressive lymphoma. Blood 138(Suppl 1), 3856 (2021).
Faude, S. et al. Absolute lymphocyte count proliferation kinetics after CAR T-cell infusion impact response and relapse. Blood Adv. 5(8), 2128–2136 (2021).
Mueller, K. T. et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130(21), 2317–2325 (2017).
Garfall, A. L. et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 3(19), 2812–2815 (2019).
Chen, P. H. et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight 5, 12 (2020).
Arcangeli, S. et al. CAR T cell manufacturing from naive/stem memory T lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J. Clin. Invest. 132(12), 3 (2022).
Biasco, L. et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat. Cancer. 2(6), 629–642 (2021).
Deng, Q. et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 26(12), 1878–1887 (2020).
Rossi, J. et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132(8), 804–814 (2018).
Acknowledgements
The authors thank the clinical medical engineers, nurses, clinical technicians and all clinicians at the Kyoto University Hospital and related hospitals as well as the patients who contributed to this research. This work was supported in part by research funding from LOTTE Foundation and the Program for the Development of Next-generation Leading Scientists with Global Insight (L-INSIGHT), sponsored by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan awarded to Y.A.
Author information
Authors and Affiliations
Contributions
Conceptualization and design of the study: F.W., T.J., and Y.A. Analysis and interpretation of data: F.W., T.J., Y.A., T.K., C.M., J.K., K.Y., M.N., and A.T.-K. Drafting or revising the manuscript: F.W., T.J., Y.A., C.M., J.K., T.K., K.Y., M.N., and A.T.-K. All authors have approved the final article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wada, F., Jo, T., Arai, Y. et al. T-cell counts in peripheral blood at leukapheresis predict responses to subsequent CAR-T cell therapy. Sci Rep 12, 18696 (2022). https://doi.org/10.1038/s41598-022-23589-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23589-9
This article is cited by
-
Quantitative PET-based biomarkers in lymphoma: getting ready for primetime
Nature Reviews Clinical Oncology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
