
Abstract
Glycine encephalopathy (GCE) is a rare autosomal recessive disorder caused by defects in the glycine cleavage complex. Here we report a patient with GCE and elevated level of glycine in both the serum and the cerebrospinal fluid. Trio-based whole-exome sequencing identified novel compound heterozygous mutations (c.738-2A>G and c.929T>C (p.Met310Thr)) in LIAS. To date, three homozygous mutations have been reported in LIAS. All previously reported GCE patients also show elevated level of serum glycine. Our data further supports LIAS mutations as a genetic cause for GCE.
Similar content being viewed by others


Introduction
Glycine encephalopathy (GCE) (MIM 605899), also known as nonketotic hyperglycinemia, is a rare autosomal recessive disorder of the glycine cleavage system1 that results in elevated glycine levels in body fluids. GCE is genetically caused by mutations in AMT (MIM 238310), GLDC (MIM 238300) or GCSH (MIM 238330), which encode T-, P- or H-protein, respectively, and together with L-protein constitute the glycine cleavage enzyme.2, 3, 4 Most GCE patients have GLDC mutations.5 Recently, GLRX5 (MIM 609588), BOLA3 (MIM 613183) and LIAS (MIM 607031) were identified as causative mutant genes for GCE.6, 7 LIAS encodes lipoic acid synthetase, a [4Fe-4S]-type iron–sulfur cluster-dependent enzyme that acts as a prosthetic group on H-protein, which is localized within the mitochondria.8 GLRX5 and BOLA3 are iron–sulfur cluster biosynthesis genes.8 All GCE patients showed elevated level of glycine in the serum, but it is only slightly increased in the cerebrospinal fluid, or even normal, in some GCE patients.6 In this study, we report a GCE individual with novel compound heterozygous mutations in LIAS.
Patients and Methods
Case report
A 21-year-old female patient was born to healthy nonconsanguineous parents. Her birth weight was 2954 g (mean), length 48.5 cm (mean) and occipitofrontal circumference 33.8 cm (75th percentile). Her initial development was normal until 18 months. Her elder sister died of herpes simplex virus encephalitis at the age of 18 months. Her elder brother is healthy.
At the age of 19 months, she suffered from an upper respiratory infection. Three days later, she vomited, became excited and then drowsy, and showed chorea-like involuntary movements. She was instantly admitted to the intensive care unit. Magnetic resonance imaging of the brain showed no abnormality (Figure 1). Her cerebrospinal fluid was examined, which showed normal results. Two weeks later, high blood glycine levels were noted (1545 nmol ml−1, normal range 179–587 nmol ml−1), which are still present (Table 1). Her cerebrospinal fluid glycine levels were also high: 45.5 nmol ml−1 (normal range 1.6–19.5 nmol ml−1; Table 1). Ketourine was not found. Three months later, magnetic resonance imaging of the brain showed high-intensity areas at the caudate nucleus and the putamen, which spread into the deep white matter (Figure 1).

T2-weighted brain magnetic resonance imaging (MRI) of the individual with LIAS mutations. (a) Examined at disease onset (19 months old). High-intensity areas are present in the caudate nucleus and the putamen. (b) Seven days after disease onset. The high-intensity areas have expanded into the white matter, especially in the frontal region. (c) One month after disease onset. High-intensity areas are noted not only in the basal nucleus but also in the subcortical white matter, especially in the frontal region. (d) At 15 years old. Owing to diffuse cerebral atrophy, the lateral ventricles have expanded. White matter atrophy is predominant in the frontal region.
At present, her height is 156 cm (25–50th percentile), weight 47.3 kg (10–25th percentile) and the occipitofrontal circumference 52.5 cm (<3rd percentile). She is now bedridden. Tube feeding has been required since 19 months of age. She speaks no meaningful words and shows no eye contact. Myoclonic movements and intractable epilepsy have been observed. Magnetic resonance imaging of the brain shows brain atrophy, especially in the white matter (Figure 1). The institutional review boards of Yokohama City University School of Medicine and Tokyo Women’s Medical University approved the study. Informed consent was obtained from the family.
Whole-exome sequencing
DNA from the patient and her parents were analyzed by whole-exome sequencing, as previously described.9 Genomic DNA was captured using the SureSelect Human All Exon v5 (50 Mb) Kit (Agilent Technologies, Santa Clara, CA, USA). Captured DNA was sequenced on a HiSeq2500 (Illumina, Inc., San Diego, CA, USA) with 101 bp paired-end reads and 7 bp index reads. Image analysis and base calling were performed by sequence control software real-time analysis and CASAVA software (v1.8) (Illumina, Inc.). Quality-controlled reads were mapped to the human reference genome (UCSC hg19, NCBI build 37.1) using Novoalign (v3.00.02; http://www.novocraft.com/products/novoalign/). After removal of PCR duplications using Picard (v1.55; http://broadinstitute.github.io/picard/), single-nucleotide variants and short insertions and deletions (Indels) were called using Genome Analysis Toolkit (v1.6-5; https://www.broadinstitute.org/gatk/) and were annotated using ANNOVAR (June 2013; http://annovar.openbioinformatics.org/en/latest/).
Prioritization of variants
All variants within exons or ±2 bp from exon–intron boundaries and registered in dbSNP137, the National Heart Lung and Blood Institute Exome Sequencing Project Exome Variant Server (NHLBI-ESP 6500), Human Genome Variation Database, or our in-house (exome data from 575 Japanese individuals) databases, as well as synonymous variants, were removed. Variants were confirmed by Sanger sequencing with an ABI PRISM 3500xl or ABI3130xl autosequencer (Life Technologies, Carlsbad, CA, USA).
Structural modeling
Crystal structure of lipoyl synthase 2 from Thermosynechococcus elongatus (PDB code 4U0P)10 was selected as the most similar model to human LIAS by Phyre2 protein fold recognition server.11
Reverse transcription-PCR and nonsense-mediated mRNA decay analysis
Lymphoblastoid cell lines derived from the patient and a healthy control were established. After incubation with 1.5 μl of dimethyl sulfoxide(as a negative control) or 1.5 μl of the protein synthesis inhibitor cycloheximide (100 mg ml−1 in dimethyl sulfoxide) (Sigma-Aldrich, St Louis, MO, USA) for 4 h. Total RNA was isolated from treated lymphoblastoid cells using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and was used for reverse transcription with the Super Script III First-Strand Synthesis System (Life Technologies). One microliter of synthesized cDNA was amplified by PCR. PCR products were electrophoresed on a 5%–20% gradient polyacrylamide gel, stained with ethidium bromide and quantitatively measured using a FluorChem 8900 (Alpha Innotech, San Leandro, CA, USA). Three independent experiments were performed. Statistical analyses were performed using analysis of variance. Each PCR band was cloned into the pCR4-TOPO vector (Life Technologies) and sequenced using ABI 3130xl autosequencer.
Results
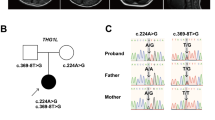
The mean read depth against RefSeq coding sequence was 89.08–123.7 reads, with 92.2%–94.7% of coding sequence covered by 20 or more reads. We focused on de novo and recessive mutations. We only found novel compound heterozygous mutations in LIAS. Sanger sequencing confirmed that c.738-2A>G was transmitted from her mother and c.929T>C (p.Met310Thr) from her father (Figure 2a and Table 1). These two mutations were not registered in dbSNP137, National Heart Lung and Blood Institute Exome Sequencing Project Exome Variant Server (NHLBI-ESP 6500), Human Genome Variation Database, or our in-house 575 Japanese control exome databases. The missense mutation (p.Met310Thr) was predicted as damaging by SIFT (http://sift.jcvi.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/), and was evolutionarily conserved (Figure 2a). Moreover, the p.Met310Thr substitution localizes to the Elp3 domain in an iron–sulfur cluster binding site predicted by SMART (http://smart.embl-heidelberg.de/) (Figure 2a). The splice site mutation (c.738-2A>G) was predicted to abolish an acceptor site by ESEfinder (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi), NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) and BDGP (http://www.fruitfly.org/seq_tools/splice.html).

All the LIAS mutations. (a) LIAS missense mutations. Novel compound heterozygous mutations (c.738-2A>G and c.929T>C (p.M310T)) identified in the patient. The upper and middle panels show the LIAS gene structure that comprises 11 exons. LIAS contains an Elp3 domain, predicted by SMART. The lower panel shows evolutionary conservation of the mutated amino acid through six different species. The altered nucleotides are highlighted in gray boxes. Three previously reported mutations (p.R249H, p.E159K and p.D215E) are highlighted in gray. (b) Mutation mapping on the crystal structure of lipoyl synthase 2 from T. elongatus (PDB code 4U0P), which is analogous to human LIAS. The mutation site is shown as sticks with translucent spheres in red in the whole (left) and the close-up (right) views of the enzyme structure. 5′-methylthioadenosine (MTA), [4Fe-4S] clusters and dithiothreitol (DTT) are depicted in color-coded sticks: green for C, red for O, blue for N, yellow for S and orange for Fe. Some hydrophobic side chains forming a core with Leu241 are shown as translucent spheres. The amino acid number in the parentheses is that for human LIAS. Black dashed lines indicate hydrogen bonds. A full color version of this figure is available at the Journal of Human Genetics journal online.
We mapped the mutation site (p.Met310Thr) on the crystal structure of lipoyl synthase 2 from T. elongatus (TeLipA2) (PDB code 4U0P),10 analogous to human LIAS (Met310 in human LIAS corresponds to Leu241 in TeLipA2), to evaluate structural impact of the mutation. The main-chain amide and carbonyl groups of Leu241 (Met310) make hydrogen bonds to adenine ring of 5′-methylthioadenosine, a breakdown product of the S-adenosylmethionine (Figure 2b), suggesting that the stability of the main-chain region around Leu241 (Met310) is important in S-adenosylmethionine binding. The side chain of Leu241 is involved in a hydrophobic core (Figure 2b) and is considered to contribute to the main-chain stability. Thus, the p.Leu241Thr (p.Met310Thr) mutation is likely to impair the enzymatic activity due to destabilized S-adenosylmethionine binding.
To examine the mutational effects of c.738-2A>G, reverse transcription-PCR was performed. Two PCR products (422-bp (isoforms 1 and 2) and 293-bp (isoform 3)) were observed in a healthy control individual regardless of the cycloheximide treatment (Figure 3a). By contrast, a 344-bp product (corresponding to isoforms 1 and 2) and a 215-bp product (corresponding to isoform 3) were newly detected from cycloheximide-treated cells from the patient at a higher level compared with untreated and dimethyl sulfoxide-treated cells, strongly indicating the abnormally spliced allele was subjected to the nonsense-mediated mRNA decay (Figure 3b). In the 422-bp product, only the missense mutation allele was seen in the electropherograms of all untreated, dimethyl sulfoxide- and cycloheximide-treated cells (Figure 3c). Therefore, the normally spliced allele only contained the missense mutation. Sequencing of the 344-bp product showed a 68-bp insertion from intron 7 and exon 8 skipping, producing a premature stop codon at amino acid position 250 (Figure 3d). In addition, sequencing of the 215-bp product confirmed a 68-bp insertion from intron 7 and exons 7 and 8 skipping, producing a premature stop codon at amino acid position 207 (Figure 3d).

A splice site LIAS mutation. (a) Reverse transcription-PCR (RT-PCR) analysis showing two PCR products (422-bp (isoforms 1 and 2) and 293-bp (isoform 3)) were observed in a healthy control individual. By contrast, a 344-bp product (corresponding to isoforms 1 and 2) and a 215-bp product (corresponding to isoform 3) were detected in cycloheximide (CHX)-treated cells from the patient at a higher level compared with untreated and dimethyl sulfoxide (DMSO)-treated cells. (b) Densitometric data of the RT-PCR products represented as mean±s.d. *P<0.05 by analysis of variance (ANOVA). NS, not significant. (c) Sequencing of the 422-bp wild type (WT) product only found missense mutation allele based on the electropherogram. (d) Schematic presentation of RT-PCR products. Sequencing of the 344-bp product showed a 68-bp insertion from intron 7 and exon 8 skipping, producing a premature stop codon at amino acid position 250. Sequencing of the 215-bp product showed a 68-bp insertion from intron 7 and exons 7 and 8 skipping, producing a premature stop codon at amino acid position 207. M, molecular size marker; WT, wild type. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
We report a family with compound heterozygous mutations (c.738-2A>G and c.929T>C (p.M310T)) in LIAS, which showed developmental delays, myoclonic movements and intractable epilepsy (Table 1). Magnetic resonance imaging of the brain showed atrophy in the white matter (Figure 1). The patient also showed late-onset involuntary movements such as chorea and elevated level of glycine in both the serum and the cerebrospinal fluid (Table 1). The splice acceptor site mutation leads to the error in splicing of intron 7 and exon 8, with a premature stop codon at amino acid position 250 in isoforms 1 and 2 or 207 in isoform 3. Consequently, the product of this mutant transcript may be subjected to nonsense-mediated mRNA decay (likely as shown in Figure 3) or produce a truncated protein at the Elp3 domain if translated (less likely).
To date, three homozygous mutations (p.R249H, p.E159K and p.D215E) have been reported in LIAS patients (Table 1).6, 7 These three missense mutations occur at evolutionarily conserved amino acids in the Elp3 domain (Figure 2a). The three reported patients show early-onset seizures within the first 2–3 days and elevated plasma glycine levels (Table 1). The patient described here shows no seizures and very late disease onset at age of 19 months (Table 1). The variable clinical features in patients with LIAS mutations may be associated with the severity of the functional impact on protein lipoylation.6
Lias homozygous knockout mice die at the early implantation stage between 7.5 and 9.5 days post coitum, suggesting that Lias is essential for embryonic development.12 The previously reported LIAS mutations are all homozygous missense mutations. Lipoamide reduction of lipoylated proteins was severely reduced in two patients (p.Arg249His and p.Glu159Lys) with early infantile lethality (at 2 and 4 years).6, 7 In contrast, one patient (p.Asp215Glu) with hypotonia and seizures showed constant development and residual lipoylated proteins, suggesting that some lipoic acid synthetase activity is retained. Thus, residual lipoic acid synthetase activity may be correlated with phenotypic severity. On the other hand, a glycine level is elevated in all patients. Thus, serum glycine levels may not be correlated with LIAS genotypes. Regardless of some biochemical and clinical data, it is difficult to extract clinical difference between our case of heterozygous mutations and reported cases of homozygous missense mutations due to the limited number of patients (n=4). Further accumulation of patients with LIAS mutations is needed.
Most GCE patients caused by GLDC or AMT mutations show hypotonia, lethargy and apnea leading to death in the first few days after birth. If they overcome apnea regaining spontaneous respiration, they develop intractable seizures and intellectual disability.13 All the four patients with LIAS mutations including our patient lived after at least 2 years of age. Therefore, LIAS mutant phenotype is relatively milder compared with the common GCE.
The acute onset of encephalopathy in our patient is interesting. We assumed that the stressful condition by the upper respiratory infection would have been the trigger of disease progression as sometimes seen in other cases of vanishing white matter.
In conclusion, we have identified for the first time, a patient with compound heterozygous missense and splice site mutations in LIAS. Further analysis of LIAS-related patients is needed to delineate the phenotypic spectrum and phenotype–genotype correlation.
References
Brenton, J. N. & Rust, R. S. Late-onset nonketotic hyperglycinemia with a heterozygous novel point mutation of the GLDC gene. Pediatr. Neurol. 50, 536–538 (2014).
Nanao, K., Okamura-Ikeda, K., Motokawa, Y., Danks, D. M., Baumgartner, E. R., Takada, G. et al. Identification of the mutations in the T-protein gene causing typical and atypical nonketotic hyperglycinemia. Hum. Genet. 93, 655–658 (1994).
Kure, S., Narisawa, K. & Tada, K. Structural and expression analyses of normal and mutant mRNA encoding glycine decarboxylase: three-base deletion in mRNA causes nonketotic hyperglycinemia. Biochem. Biophys. Res. Commun. 174, 1176–1182 (1991).
Koyata, H. & Hiraga, K. The glycine cleavage system: structure of a cDNA encoding human H-protein, and partial characterization of its gene in patients with hyperglycinemias. Am. J. Hum. Genet. 48, 351–361 (1991).
Kanekar, S. & Byler, D. Characteristic MRI findings in neonatal nonketotic hyperglycinemia due to sequence changes in GLDC gene encoding the enzyme glycine decarboxylase. Metab. Brain Dis. 28, 717–720 (2013).
Baker, P. R. 2nd, Friederich, M. W., Swanson, M. A., Shaikh, T., Bhattacharya, K., Scharer, G. H. et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137, 366–379 (2014).
Mayr, J. A., Zimmermann, F. A., Fauth, C., Bergheim, C., Meierhofer, D., Radmayr, D. et al. Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am. J. Hum. Genet. 89, 792–797 (2011).
Mayr, J. A., Feichtinger, R. G., Tort, F., Ribes, A. & Sperl, W. Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 37, 553–563 (2014).
Tsurusaki, Y., Koshimizu, E., Ohashi, H., Phadke, S., Kou, I., Shiina, M. et al. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat. Commun. 5, 4011 (2014).
Harmer, J. E., Hiscox, M. J., Dinis, P. C., Fox, S. J., Iliopoulos, A., Hussey, J. E. et al. Structures of lipoyl synthase reveal a compact active site for controlling sequential sulfur insertion reactions. Biochem. J. 464, 123–133 (2014).
Kelley, L. A. & Sternberg, M. J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 (2009).
Yi, X. & Maeda, N. Endogenous production of lipoic acid is essential for mouse development. Mol. Cell. Biol. 25, 8387–8392 (2005).
Hennermann, J. B., Berger, J. M., Grieben, U., Scharer, G. & Van Hove, J. L. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J. Inherit. Metab. Dis. 35, 253–261 (2012).
Acknowledgements
We thank the patients and their families for their participation in this study. We also thank Nobuko Watanabe for her excellent technical assistance. This work was supported by the Ministry of Health, Labour and Welfare of Japan; the Japan Society for the Promotion of Science (Grants-in-Aid for Scientific Research (A) (B) (C)); the Takeda Science Foundation; the Japan Science and Technology Agency (the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems); the Ministry of Education, Culture, Sports, Science and Technology of Japan (the Strategic Research Program for Brain Sciences); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Tsurusaki, Y., Tanaka, R., Shimada, S. et al. Novel compound heterozygous LIAS mutations cause glycine encephalopathy. J Hum Genet 60, 631–635 (2015). https://doi.org/10.1038/jhg.2015.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.72
This article is cited by
-
Pharmacological Aspects of the Use of Lipoic Acid (Review)
Pharmaceutical Chemistry Journal (2022)
-
TMEM67 mutations found in a case of Joubert syndrome with renal hypodysplasia
CEN Case Reports (2016)
