
Abstract
Diaphanospondylodysostosis (DSD), caused by loss of bone morphogenetic protein-binding endothelial regulator (BMPER), has been considered a lethal skeletal dysplasia characterized by severe deficiency of vertebral body and sacral ossification, reduced rib number and cystic kidneys. In this study, however, we have demonstrated that variants in BMPER may cause a milder disorder, without renal anomalies, that is compatible with long-term survival. Four siblings, three males and one female, presented with severe congenital scoliosis associated with rib and vertebral malformations as well as strikingly delayed ossification of the pedicles. The female was stillborn from an unrelated cause. Stabilization of the scoliosis with expandable titanium rods was successful in the three boys, all of whom have short stature. An autosomal recessive mode of inheritance was hypothesized. Single nucleotide polymorphism microarray analysis was performed for three of the siblings to identify autosomal genes with shared allele patterns, suggesting possible linkage. Exome sequencing of one sibling was then performed. Rare variants were identified in 347 genes with shared alleles. Only one of these genes had bi-allelic variants in a gene strongly expressed in paraxial mesenchyme: BMPER, which is the cause of DSD, an autosomal recessive disorder. The disorder described herein could represent an attenuated form of DSD or could be designated a separate entity such as spondylopedicular dysplasia.
Similar content being viewed by others

Introduction
We evaluated a set of four siblings who presented with severe progressive congenital scoliosis, accompanied by short stature, delayed ossification and hypoplasia of the vertebral pedicles, minor vertebral defects, and decreased number of ribs. Congenital scoliosis (estimated incidence: 1 in 2000)1 may result from an isolated malformation (~50%), such as, for example, hemivertebrae, or as part of a more complex disorder including several of the skeletal dysplasias.2, 3 The spondylocostal or spondylothoracic dysostoses were considered unlikely given the relative paucity of segmentation defects. Another entity on the differential diagnosis was diaphanospondylodysostosis (DSD), an extremely rare autosomal recessive skeletal dysplasia with fewer than 20 affected individuals described. Disturbed ossification and morphogenesis of the axial skeleton, multicystic kidneys with nephroblastomatosis, and occasional cleft palate and/or mild craniofacial features comprise the phenotypic effects of this syndrome.4 The causative gene, BMPER (bone morphogenetic protein (BMP)-binding endothelial regulator), regulates BMP signaling, and thereby influences angiogenesis, adipogenesis and osteoblast differentiation among other developmental processes.5, 6, 7 Initially, DSD was also considered unlikely because of the lack of nephroblastomatosis and lethal thoracic hypoplasia in these siblings. Thus, a novel autosomal recessive skeletal dysplasia was hypothesized and the underlying causative gene was sought through a combined linkage and exome sequencing approach.
Patients and Methods
Clinical phenotyping
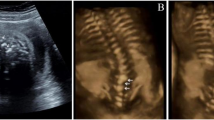
Written, informed consent for this study was obtained per an institutional review board approved protocol (H09-00230). The first child, a female, was stillborn at term as a result of a probable umbilical cord accident. In addition to findings in keeping with asphyxiation, an autopsy demonstrated bilateral cleft palate, micrognathia, absent twelfth ribs bilaterally, coronal clefts in multiple vertebra and under-ossified, hypoplastic pedicles in the thoracic region (Figure 1a). Histopathology of a vertebra and rib was normal (Supplementary Figure 1). A male sibling was born with thoracolumbar (T7 to L4) scoliosis, lack of thoracic pedicle ossification, 10 ribs on the right side and 11 on the left and an L2 hemivertebra. The third sibling, a male, had mid-thoracic (T4–T8) dextroscoliosis, 11 ribs on the right, a single rib fusion, several fused pseudoarticulation abnormalities and absent vertebral pedicle ossification in the thoracic spine (Figure 1). By age 4 years, ossification of the lower thoracic pedicles could be observed. During the pregnancy with the fourth sibling, a second trimester ultrasound showed scoliosis. Shortly after birth he was found to have 10 rib pairs, mid-thoracic kyphoscoliosis and delayed ossification of the mid-thoracic and lower lumbar pedicles, which were also hypoplastic. All three boys have undergone vertical expandable prosthetic titanium rib placement with successful stabilization of their scoliosis. Their weight and height parameters have tracked at less than the first percentile (sib 2: −4.7 s.d., sib 3: −3.8 s.d., sib 4: −5.0 s.d.); armspan is also below the first percentile (sib 2: −3.9 s.d., sib 3: −3.4 s.d., sib 4: −4.0 s.d.) indicating impairment of linear growth irrespective of the spinal deformity. Growth hormone levels in the living siblings were in the normal range. None of the siblings have had tracheomalacia nor any other issues with their upper or lower airways. They all have required tympanostomy tubes for middle ear fluid accumulation. Sibling 2 has celiac disease with a positive elevation of tissue transglutaminase. Cognitive development has been normal with above-average academic performances. There are no other siblings in the family.

(a) Chest radiograph of Sibling 1 demonstrating lack of ossification of the pedicles (arrowhead), particularly in the upper thoracic spine. The twelfth rib pair is missing. (b) A radiograph of Sibling 2’s pelvis demonstrates decreased ossification of the sacrum, posterior fusion defects and large ossification defects of the ischia (arrowhead). (c,d) show the progression of rotatory scoliosis between birth (c) and 2 years of age (d) in Sibling 3. A three-dimensional computed tomography (CT) display of the thoracic spine of Sibling 3 (e) at 12 months demonstrates rotation of the spine as well as absent ossification of the pedicles (arrowhead). A full color version of this figure is available at the Journal of Human Genetics journal online.
The parents are unrelated to one another and are both of British-European descent. The father has absence of the right kidney and multiple parapelvic macrocysts in the left kidney. The unilateral renal aplasia is a finding shared with his likely monozygotic twin. The father also has very mild (<5°) thoracic scoliosis and mild anterior wedging of T12 and L1. The paternal grandmother has 11 pairs of ribs. The mother has no rib anomalies and a thoracic curve <5°, and her sister reportedly had adolescent idiopathic scoliosis. The mother has not had a renal ultrasound.
SNP genotyping
DNA was extracted from peripheral blood lymphocytes from Siblings 2–4, and from frozen spleen from Sibling 1. The DNA from Sibling 1 was degraded, and therefore amplification was undertaken using the Whole Genome Amplification kit (Sigma-Aldrich, St. Louis, MO, USA) as per the manufacturer instructions. The siblings’ samples were then hybridized to the Affymetrix 6.0 single nucleotide polymorphism (SNP) array comprising 1.8 million probes. Data from the scanned array were analyzed using Genotype Console 2.1 (Affymetrix, Santa Clara, CA, USA). Greater than 93% of SNPs passed quality control thresholds for Siblings 2–4.
Exome sequencing
Exome sequencing was carried out for Sibling 4 (after random selection among the siblings). Agilent SureSelect 38 Mb (Santa Clara, CA, USA) was used for exon capture. Two lanes on an Illumina Genome Analyzer II flow cell (Illumina, San Diego, CA, USA) were used to generate 132 million paired-end 75 bp reads. Alignment and variant calling was performed with Nextgene software using default settings (SoftGenetics, State College, PA, USA). Variants were filtered for presence in coding exons ±20 bp intronic sequence and a population frequency of <0.1%, according to dbSNP 135 and the Exome Variant Server.
Genotyping of additional controls
A restriction digest of PCR amplification products was used to genotype 192 control individuals of European descent for the c.251G>T variant, following the manufacturer’s recommended protocol for Bsp1286 I (NEB). PCR amplification and Sanger sequencing genotyped the same controls for the c.1078+5G>A splice variant. Control samples were obtained from Sigma-Aldrich.
Splicing analysis
Primary dermal fibroblast cultures were established from skin biopsies from the father and Sibling 3. RNA was extracted using RNeasy Mini Kit (Qiagen, Redwood City, CA, USA), and complimentary DNA was synthesized directly from RNA with Superscript III Reverse Transcriptase (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. Exons 11–13 were PCR amplified (using primers: 5′-GTGCTTGTGTGAAAGGCAGG-3′; 5′-AAACGTACTGACACGTCCCC-3′), and the resultant bands were then gel extracted. The amplicons were cloned into TOP10 competent Escherichia coli with using the TA Cloning Kit and pCR 2.1 Vector (Life Technologies). Sanger sequencing of plasmids was performed using M13 universal primers.
Results
Autosomal SNPs were analyzed for minimum 25 kb segments (containing at least 10 SNPs) of identical haplotypes among Siblings 2–4. There were 4266 such shared segments covering 3426 genes, although for many of these, the SNPs were entirely homozygous, and it was uncertain how many segments were indicating true identity by descent. Of these genes, 98 were identified as strongly expressed in paraxial mesenchyme, as documented in the Mouse Genome Informatics database (www.informatics.jax.org). Paired-end read exome sequencing of Sibling 4 generated at least 10-fold coverage for 87.2% of the targeted regions. A search for genes known to cause skeletal dysplasia harboring rare non-synonymous variants, either homozygous or potentially compound heterozygous, revealed only BMPER. SOX9 was specifically assessed to rule out any sequencing variants and to ensure all exons were adequately covered. Of the 98 potentially linked paraxial mesenchymal genes, only three were found to have rare variants by exome sequencing: BMP2K, HOXA7 and BMPER. Of these three, BMP2K had only one variant, which was synonymous, and thus was not considered further. HOXA7 had one moderate confidence variant and possible additional low confidence variants, however, Sanger sequencing of the entire HOXA7 coding region did not confirm any true variants. BMPER showed two rare variants (described relative to ENST00000297161; NM_133468.4): c.251G>T (p.C84F) and c.1078+5G>A. Subsequent Sanger sequencing in the family showed the father carried the splice-site variant (Supplementary Figure 2), the mother carried the missense variant, and the four siblings each had inherited both variants. The missense variant was not present in dbSNP v.143 or ExAC, and the splice-site mutation of the fifth nucleotide of intron 12 was reported once in dbSNP (rs559550154), being heterozygously present once in a cohort of 1500 individuals, and was absent in ExAc. Using assays optimized to detect these variants, neither mutation was detected in 192 anonymous control samples of the same Northern European ethnic origin as the family. The missense variant was predicted by MutationTaster to be disease-causing at a probability of 0.999 and the amino acid is conserved throughout vertebrates.8 The splicing variant was predicted to be disease-causing at a probability of 0.996, and the nucleotide is conserved across vertebrate species. PCR amplification of complimentary DNA generated from fibroblast RNA from the father and Sibling 3 demonstrated the presence of an extra, aberrant band, which was then cloned into a vector. Sanger sequencing subsequently confirmed that BMPER exon 12 was removed during mRNA splicing (Figure 2). BMPER is a secreted extracellular protein of the chordin family containing a trypsin inhibitor-like cysteine rich domain (TIL), a von Willibrand factor type D domain, and five von Willibrand factor type C domains (VWC1), the first of which avidly binds BMP2, according to X-ray crystallography, and can compete against BMP receptor types I and II with much higher affinity for BMP than the other VWC domains.9, 10 The C84 residue is located in subdomain 2 of VWC1. This cysteine residue forms a disulfide bond with C102, also a highly conserved residue, adding rigidity to VWC1’s rod-like secondary structure.9 Furthermore, subdomain 2 of VWC1 binds chordin, a BMP inhibitor, and could thereby potentially contribute a pro-BMP signaling effect.11 Splicing out of exon 12 leads to a frameshift, introducing a premature stop codon 30 amino acids downstream, with resultant loss of the von Willebrand factor type D domain and the trypsin inhibitory-like cysteine rich domain. The fact that some aberrant mRNA was detected suggests that a truncated protein may be produced, and hence the allele may by hypomorphic. The missing carboxy-terminal von Willebrand factor type D domain contains a heparin binding site that binds to cell surface heparan sulfate proteoglycans, thereby limiting BMPER diffusion.12, 13

(a) Schematic of BMPER protein structure. Locations of the two mutations are starred. This 685 amino acid protein contains five von Willibrand factor type C domains, a von Willibrand factor type D domain and a trypsin inhibitor-like cysteine rich domain. (b) Chromatogram from exons 11 through 13 from Sibling 3’s fibroblast complimentary DNA, which was sequenced and gel extracted after cloning of a PCR product. Identical sequence was also obtained from the father’s fibroblast culture (data not shown). The schematic demonstrates the loss of exon 12 in its entirety. The mutation resides in the consensus splice donor of intron 12. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
We investigated four siblings who presented with a very severe, progressive congenital rotatory scoliosis requiring surgical stabilization because of thoracic volume loss leading to respiratory compromise. To identify the underlying causative gene, we performed exome sequencing looking for very rare or novel pathogenic mutations by filtering for (1) genes expressed in the developing spinal column (paraxial mesenchyme); (2) variants in genes that showed SNP patterns shared by all four siblings and (3) genes with two variants, in keeping with an autosomal recessive model. Using this approach, we identified bi-allelic BMPER mutations in this family, which expands the phenotypic spectrum of BMPER-related disorders to include a non-lethal, milder skeletal phenotype without renal anomalies. In parallel, we also searched for variants in known human skeletal disorders and arrived only at the same variants in BMPER. Despite bioinformatically arriving at only a single gene, it is of course possible that a different, novel gene or occult variants in a known gene could be responsible, but the strong phenotypic similarities with DSD argue for causation.
DSD, caused by recessive mutations in BMPER (also known as crossveinless 2 for its role in forming veins in the wing of drosophila),14 was considered early in the differential diagnosis, but was considered unlikely because of several key differences in this family compared with the established DSD phenotype. To date, ∼20 individuals with DSD have been reported. Early childhood lethality has been reported in all previous patients, but has not occurred in this family. Decreased ossification is much more pronounced in the vertebral bodies, whereas in this family, unossified pedicles were much more striking. The kidneys are usually cystic with nephrogenic rests, but this phenotype was not seen in our affected sibship.15 Segmentation defects and missing ribs are other recurrent features in the syndrome, and at least the latter was present in these siblings. Whereas the histopathology of rib and vertebrae from previous reported severely affected patients has shown incomplete ossification and gaps in cartilage formation,15 this was not obvious from the studies on Sibling 1 in this family. There is some overlap of craniofacial features between our family and others; we observed short neck, mild micrognathia and short nose, but no hypertelorism or low-set ears as previously reported. Clefting of the palate, present in Sibling 1, has been noted in one other individual, and has also been observed in the null mouse model. The null Bmper mouse dies at birth, showing a short trunk, short tail, small size, missing ribs, lack of tracheal cartilage, decreased axial ossification and small kidneys, but the heterozygotes survive with subtler anomalies, such as abnormal alveolar development and delayed branching in the lungs.16
Considering the finding of bi-allelic mutations in BMPER in these siblings, the diagnosis of attenuated DSD seems appropriate. It has been proposed that a milder form of DSD may have been already described under the name ischiospinal dysostosis, which is a syndrome characterized by short stature, poor ossification of ischial rami, sacral hypoplasia, segmental vertebral anomalies, scoliosis, nephroblastomatosis and cystic kidneys.15, 17, 18 Sequencing of BMPER in any individuals with this diagnosis has not yet been reported. With the exception of obvious nephroblastomatosis, the siblings in this paper do show the major findings of ischiospinal dysostosis, including ossification defects of the ischia. The milder phenotype may relate to the impact of these particular mutations on the protein function or may be attenuated because of unknown modifiers. Considering the rather uniform presentation among these siblings, the former possibility seems more likely. The oldest survivor of DSD previously reported lived to age 5 years, succumbing to a relapse of nephroblastoma.19 The oldest sibling in this report is now 13 years old and doing well. These siblings all had normal renal ultrasound findings, however we cannot be certain about the absence of nephrogenic rests. The youngest sibling in this family is now 6 years old, nearing the end of the period of high risk for nephroblastoma. Seven other BMPER mutations have been reported thus far in individuals with DSD, comprising four nonsense substitutions, one frame-shifting deletion with n-terminal truncation, one splice-site variant of unknown effect, and one missense variant in von Willibrand factor type D domain.14, 20 Potential protein products resulting from these variants have not been studied, and although most of these might be expected to have null effect, this is not certain. Truncations occur more proximally in the previously reported individuals, who are more severely affected, whereas the frame-shifting variant in this family is closer to the C terminus. Furthermore, the second allele in our family is missense, possibly only affecting secondary structure, supporting our hypothesis that the type of mutation correlates with disease severity.
DSD is notable for featuring both abnormal segmentation and ossification. The term dysostosis highlights the anomalous morphogenesis, but the ongoing perturbation of bone growth and ossification evident in these siblings from their severe linear growth restriction indicates that there is both dysostosis as well as dysplasia. The precise mechanism of the scoliosis in these siblings is not clear, but several developmental anomalies are present in the spine including striking delay in the ossification of the vertebral pedicles. Congenital scoliosis most often results from mechanical imbalance due to asymmetrical vertebral malformation,21 but can also result from general lack of structural integrity of the vertebral column. The spine develops from paraxial mesoderm via a process of somite formation and patterning through Notch, wnt, TGF-β and FGF signaling pathways.22, 23 BMPs, part of the transforming growth factor-β superfamily, are required throughout this process from very early on; for example, BMP2 and BMP4 production in the overlying surface ectoderm induces the dorsal elements of the vertebrae.24 BMPER avidly binds and modulates BMPs 2, 4, 6 and 7, and has a key role in axial skeletal formation and patterning through both activating and inhibiting roles.25 BMPER expression increases concordantly with BMP gene expression during development.13
It is interesting to note that the father has mild scoliosis and a single kidney with macrocysts. His twin brother also has unilateral renal agenesis, and, based on identical physical appearance judged by a clinical geneticist, is likely monozygous. Familial tendencies have been recognized in idiopathic scoliosis and susceptibility factors are being sought.26 Further studies could explore possible physiological effects of BMPER mutations in heterozygote carriers.
In summary, we describe an attenuated form of DSD due to BMPER mutations presenting with severe scoliosis, clefting of the palate, missing ribs and delayed ossification of the pedicles, but notably lacking previously well recognized features of this condition, including cystic renal dysplasia and early lethality. For these siblings, severe progressive infantile scoliosis has been the most striking clinical finding, creating further clinical distinction from other patients. Hence, we have considered whether a separate designation should be given to this phenotype, such as spondylopedicular dysplasia, but likely the term attenuated DSD is sufficient. The differential diagnosis includes acampomelic campomelic dysplasia, particularly when ossification defects of the vertebral pedicles are present, and analysis of SOX9 facilitates differentiation. Another possible diagnosis is ischiospinal dysostosis, for which the genetic basis has not yet been determined, and which has been considered as part of the phenotypic range of DSD. The study of further children with BMPER-related skeletal disease will help delineate the full phenotypic spectrum and allow for the appropriate decisions around ‘lumping versus splitting’ in the classification of these disorders.
References
Wynne-Davies, R. Congenital vertebral anomalies: aetiology and relationship to spina bifida cystica. J. Med. Genet. 12, 280–288 (1975).
Jaskwhich, D., Ali, R. M., Patel, T. C. & Green, D. W. Congenital scoliosis. Curr. Opin. Pediatr. 12, 61–66 (2000).
Erol, B., Tracy, M. R., Dormans, J. P., Zackai, E. H., Maisenbacher, M. S., O’Brien, B. A. et al. Congenital scoliosis and vertebral malformations: characterization of segmental defects for genetic analysis. J. Pediatr. Orthop. 24, 674–682 (2004).
Prefumo, F., Homfray, T., Jeffrey, I., Moore, I. & Thilaganathan, B. A newly recognized autosomal recessive syndrome with abnormal vertebral ossification, rib abnormalities, and nephrogenic rests. Am. J. Med. Genet. A 120A, 386–388 (2003).
Heinke, J., Wehofsits, L., Zhou, Q., Zoeller, C., Baar, K., Helbing, T. et al. BMPER is an endothelial cell regulator and controls bone morphogenetic protein-4-dependent angiogenesis. Circ. Res. 103, 804–812 (2008).
Binnerts, M. E., Wen, X., Cante-Barrett, K., Bright, J., Chen, H., Asundi, V. et al. Human Crossveinless-2 is a novel inhibitor of bone morphogenetic proteins. Biochem. Biophys. Res. Commun. 315, 272–280 (2004).
Liu, Z., Sun, W., Zhao, Y., Xu, C., Fu, Y., Li, Y. et al. The effect of variants in the promoter of BMPER on the intramuscular fat deposition in longissimus dorsi muscle of pigs. Gene 542, 168–172 (2014).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Zhang, J.-L., Qiu, L. Y., Kotzsch, A., Weidauer, S., Patterson, L., Hammerschmidt, M. et al. Crystal structure analysis reveals how the Chordin family member crossveinless 2 blocks BMP-2 receptor binding. Dev. Cell 14, 739–750 (2008).
Fiebig, J. E., Weidauer, S. E., Qiu, L. -Y., Bauer, M., Schmieder, P., Beerbaum, M. et al. The clip-segment of the von Willebrand domain 1 of the BMP modulator protein Crossveinless 2 is preformed. Molecules 18, 11658–11682 (2013).
Zhang, J. -L., Patterson, L. J., Qiu, L.-Y., Graziussi, D., Sebald, W. & Hammerschmidt, M. Binding between Crossveinless-2 and Chordin von Willebrand factor type C domains promotes BMP signaling by blocking Chordin activity. PLoS ONE 5, e12846 (2010).
Rentzsch, F., Anton, R., Saina, M. & Hammerschmidt, M. Asymmetric expression of the BMP antagonists chordin and gremlin in the sea anemone Nematostella vectensis: Implications for the evolution of axial patterning. Dev. Biol. 296, 375–387 (2006).
Serpe, M., Umulis, D., Ralston, A., Chen, J. & Olson, D. J. The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Dev. Cell 14, 940–953 (2008).
Funari, V. A., Krakow, D., Nevarez, L., Chen, Z., Funari, T. L., Vatanavicharn, N. et al. BMPER mutation in diaphanospondylodysostosis identified by ancestral autozygosity mapping and targeted high-throughput sequencing. Am. J. Hum. Genet. 87, 532–537 (2010).
Vatanavicharn, N., Graham, J. M., Curry, C. J., Pepkowitz, S., Lachman, R. S., Rimoin, D. L. et al. Diaphanospondylodysostosis: six new cases and exclusion of the candidate genes, PAX1 and MEOX1. Am. J. Med. Genet. 143A, 2292–2302 (2007).
Kelley, R., Ren, R., Pi, X., Wu, Y., Moreno, I., Willis, M. et al. A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J. Cell Biol. 184, 597–609 (2009).
Nishimura, G., Kim, O. H., Sato, S. & Hasegawa, T. Ischiospinal dysostosis with cystic kidney disease: report of two cases. Clin. Dysmorphol. 12, 101–104 (2003).
Spranger, J., Self, S., Clarkson, K. B. & Pai, G. S. Ischiospinal dysostosis with rib gaps and nephroblastomatosis. Clin. Dysmorphol. 10, 19–23 (2001).
Scottoline, B., Rosenthal, S., Keisari, R., Kirpekar, R., Angell, C. & Wallerstein, R. Long-term survival with diaphanospondylodysostosis (DSD): survival to 5 years and further phenotypic characteristics. Am. J. Med. Genet. 158A, 1447–1451 (2012).
Ben-Neriah, Z., Michaelson-Cohen, R., Inbar-Feigenberg, M., Nadjari, M., Zeligson, S., Shaag, A. et al. A deleterious founder mutation in the BMPER gene causes diaphanospondylodysostosis (DSD). Am. J. Med. Genet. 155, 2801–2806 (2011).
Oskouian, R. J., Sansur, C. A. & Shaffrey, C. I. Congenital abnormalities of the thoracic and lumbar spine. Neurosurg. Clin. N. Am. 18, 479–498 (2007).
Turnpenny, P. D., Alman, B., Cornier, A. S., Giampietro, P. F., Offiah, A., Tassy, O. et al. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev. Dyn. 236, 1456–1474 (2007).
Kusumi, K. & Dunwoodie, S. L. (eds). The Genetics and Development of Scoliosis. (Springer, New York, NY, USA, 2010).
Watanabe, Y., Duprez, D., Monsoro-Burq, A. H., Vincent, C. & Le Douarin, N. M. Two domains in vertebral development: antagonistic regulation by SHH and BMP4 proteins. Development 125, 2631–2639 (1998).
Ikeya, M., Kawada, M., Kiyonari, H., Sasai, N., Nakao, K., Furuta, Y. et al. Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development 133, 4463–4473 (2006).
Ogilvie, J. W. Update on prognostic genetic testing in adolescent idiopathic scoliosis (AIS). J. Pediatr. Orthop. 31, S46–S48 (2011).
Acknowledgements
SNP microarray genotyping was performed at The Centre for Applied Genomics (University of Toronto, Canada). This work was supported by a grant from the Rare Disease Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Zong, Z., Tees, S., Miyanji, F. et al. BMPER variants associated with a novel, attenuated subtype of diaphanospondylodysostosis. J Hum Genet 60, 743–747 (2015). https://doi.org/10.1038/jhg.2015.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.116
This article is cited by
-
Cytogenomic aberrations in isolated multicystic dysplastic kidney in children
Pediatric Research (2022)
-
Extending the phenotype of BMPER-related skeletal dysplasias to ischiospinal dysostosis
Orphanet Journal of Rare Diseases (2016)

{kind=link}
{kind=link}