
Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder characterized by multiple hamartias and hamartomas involving throughout the body. To date, many TSC1 and TSC2 mutations have been reported all over the world, however, few TSC mutation studies have been performed in the Japanese population, and genetic characteristics of Japanese TSC patients are not yet clear. In this study, we analyzed TSC1 and TSC2 in 57 Japanese patients with TSC (8 familial and 49 sporadic; 46 definite and 11 suspect TSC) and identified 31 mutations including 11 TSC1 mutations (two familial and nine sporadic; all definite TSC) and 20 TSC2 mutations (2 familial and 18 sporadic; 19 definite and 1 suspect TSC). We also reviewed all Japanese TSC mutations previously reported. Our study demonstrates significantly higher incidence (P=0.007) of TSC1 mutations among sporadic TSC patients in the Japanese population compared with US and European studies. No differences emerged in mutation distributions and types in precedent studies, excepting low frequency of the TSC2 nonsense mutation. Comparing clinical manifestations, developmental delay and/or mental retardation were milder in TSC1 patients than TSC2 patients for its frequency (P=0.032) and severity (P=0.015); however, no other symptoms were clearly different.
Similar content being viewed by others
Introduction
Tuberous sclerosis complex (TSC) is a variably expressed multisystem disorder. It manifests lesions with misarranged undifferentiated dysplastic cells. These dysplastic cells do not grow more rapidly than normal cells in some lesions (called hamartias), but in other lesions, grow into benign tumors (called hamartomas) or rarely grow into malignant tumors such as renal cell carcinoma.1 As is well known, characteristic hamartias of TSC are hypomelanotic skin macules and brain cortical tubers, whose disease name is derived from macroscopic pathological brain feature of cortical tubers described by Bourneville as ‘tuberous sclerosis of the cerebral convolutions’.2 Also characteristic hamartomas are subependymal nodules, facial angiofibromas, renal angiomyolipomas (AML) and cardiac rhabdomyomas. In practice, however, entire body organs are involved in patients with TSC, including retina, lung, liver, pancreas, intestines, endocrine system, bone and teeth,1 and established clinical diagnostic criteria is widely used.3 All hamartias are embryonic lesions, whereas hamartomas appear in an age-dependent fashion; that is, cardiac rhabdomyomas grow in the fetus through early infanthood then regress, facial angiofibromas become apparent in puberty, renal AML emerge in adults and subungual fibromas present after middle age.4 At least half of the patients with TSC have developmental delay or mental retardation, sometime with intractable epilepsy, though the spectrum of severity in individual patients varies widely between families or even within a family.1
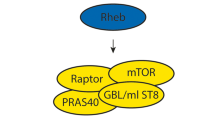
TSC is an autosomal dominantly inherited disorder, and its estimated frequency in the population is about 1 in 6000 of whom approximately two-thirds are sporadic and the remaining segregating in families.5 The disease is caused by the mutation of one of two tumor suppressor genes, TSC1 (OMIM # 191100) and TSC2 (OMIM #191092), which encode hamartin and tuberin, respectively.6, 7 These proteins form a functional heterodimer, and this intracellular complex (TSC complex) is a universal sensor of several intracellular and extracellular signals; for example, growth factor stimulation, hypoxia and energy levels.8 The most remarkable function of the TSC complex is GTPase activating protein activity for Ras homolog enriched in brain (Rheb), which converts Rheb–GTP to Rheb–GDP. Since Rheb–GTP activates the mammalian target of rapamycin (mTOR), in cells lacking proper function of the TSC complex there are increased levels of Rheb–GTP, which leads to activation of mTOR, causing increased cell size and growth through phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1, ribosomal protein S6 kinase beta-1 and eukaryotic translation initiation factor 2 (eEF-2K).8, 9
Concordant with tumor suppressor function, loss of heterozygosity (LOH) of both TSC1 and TSC2 loci have been demonstrated in TSC lesions; however, reported LOH frequencies differed depending on the genes and tumor types. LOH was demonstrated more frequently in TSC2 (37%) than TSC1 (6%), with the highest frequency observed in renal AMLs (56%), intermediate rates observed in subependymal giant cell astrocytoma (SEGA; 39%), skin lesions (33%) and cardiac rhabdomyoma (23%), and the lowest rates observed in cortical tubers (15%).10 However, increased phosphorylated isoforms of S6 kinase beta-1 and 4E-binding protein 1, which exhibit hyperactive mTOR signaling, was shown in cortical tubers from patients with TSC.11 Also, abnormal phosphorylation of tuberin in cortical tubers, which induced post-translational inactivation of the TSC2 product without second hit on DNA level, was demonstrated.12 Some investigators postulated that this modification occurs through activation of extracellular signal-regulated kinase (ERK).8, 13 More recently, hamartin and tuberin have been discovered to bind to at least 40 additional proteins, and thus there are numerous potentials and yet undefined effects of TSC gene mutations.14
To date, there are several large cohort studies for TSC mutations in US,15, 16, 17 UK,18 Denmark,19 Netherland,20 Australia,21 Taiwan22 and Korea.23, 24 Through these studies, several aspects of the molecular genetics of TSC are becoming apparent. Overall mutation distribution among patients with TSC shows higher frequency of TSC2 mutations than TSC1 mutations, with equally represented TSC1 and TSC2 mutations in familial TSC, but TSC2 mutations are more common than TSC1 mutations in sporadic cases.15 Although all types of mutations including large deletions and point mutations have been identified in TSC2; the majority of TSC1 mutations are frameshift, nonsense and splicing mutations, but missense mutations and large deletions are rare.16 There are no mutational hot spots in either TSC1 or TSC2. The type of TSC1 or TSC2 mutation (protein truncation vs missense) and the location of the mutation do not predict the severity of the phenotype. The exception is that some TSC2 missense mutations (that is, p.R905Q and p.Q1503P) are associated with milder disease phenotypes,25, 26 and TSC2/PKD1 contiguous gene deletion is correlated with polycystic kidney disease.27 Detailed genotype/phenotype data indicated that TSC2 mutations produce a more severe phenotype than TSC1 mutations especially in intellectual disability.16, 17
Although large amounts of data on TSC mutations and phenotypes have been reported worldwide, in Japan only three TSC mutation cohort studies, including our previous study,28, 29, 30 and three TSC mutations collected reports have been presented.31, 32, 33 The diversity of mutations, phenotype characteristics and genotype correlations in the Japanese TSC population are not yet clear. In this study, we report a mutational analysis of both TSC1 and TSC2 genes in 57 unbiased sequential Japanese patients with TSC. We screened whole-coding exons of both genes and performed long PCR and Southern blotting for detecting large gene deletions. Also, detailed clinical data were corrected by symptom check lists for each patient. In addition, we reviewed and verified all previously reported Japanese TSC mutations available in the literature and summarized them in a unified format and compared them to TSC mutations reported in other races.
Materials and methods
Patient collection and clinical evaluations
We analyzed 57 Japanese patients with TSC, 8 familial and 49 sporadic cases, collected between 2002 and 2011. These patients were clinically diagnosed according to established criteria,3 and 46 patients diagnosed as definite TSC and 11 patients as suspect TSC. Of the 57 patients, 15 patients were referred to our department in Kanazawa University Hospital or Kanazawa Medical University Hospital. These patients were clinically evaluated by us with the clinical data check list, according to Dabora et al.,16 and corrected DNA samples. The remaining 42 samples were collected from regions all over Japan in response to requests for genetic diagnosis of TSC with the same clinical data check list filled out by the primary doctors. The clinical data check list not only assesses TSC diagnosis, but also scores severity of the patients’ phenotype in several aspects. In the original check list, mental retardation, facial angiofibroma, renal AML and liver AML are scored as grade 0–3, and renal cysts are scored as grade 0–4 depending on severity. Actual numbers of subependymal nodules and cortical tubers are counted on brain imaging (computed tomography or magnetic resonance).
In this study, we modified the original check list at two points for convenience. First, for the assessment of mental retardation, the original check list uses speech and reading abilities as applied to only patients 8 years of age or older. Our subjects included many infants and toddlers and we could not use this scale for these young patients. Thus, we defined new criteria for ‘Developmental delay and/or mental retardation’ for our patients as follows. Grade 0: intercultural quotient or developmental quotient evaluated in any established scale is 70 or more and no obvious psychomotor developmental delay was observed in infants and toddlers by a pediatric neurologist; or, grade 0 in the mental retardation item in the original check list for older children and adults, corresponding to reading ability above a third grade level (8 years of age). Grade 1: intercultural quotient or developmental quotient is between 50 and 69, and pediatric neurologist observed mild developmental delay, or grade 1 in the original check list, corresponding to reading at less than a third grade level, in older children and adults. Grade 2: intercultural quotient or developmental quotient is <50, and pediatric neurologist observed substantial developmental delay, or grade 2 in the original check list in older children and adults, corresponding to some speech ability, no reading ability or grade 3 in the original check list in older children and adults, no speech ability. We eliminated five patients <6 months of age who were too young to evaluate (family 12, 26, 31, 32 and 47).
Second, the number of subependymal nodules and cortical tubers are difficult to count precisely. It is dependent on performance of the imaging machine, the condition of the slice to the region and any body motion of the patients. So, we simply scored as grade 0: no lesion, grade 1: single lesion and grade 2: multiple lesions.
The study was approved by the institutional review board and all samples were obtained with informed consent.
Mutational screening
Genomic DNA was extracted from whole blood by rapid methods.34 DNA samples obtained from 50 normal volunteer individuals (medical students of Kanazawa University) were used as a control panel.
We analyzed whole-coding exons of both TSC1 and TSC2 genes. For the first 10 patients (including nine definite and one suspect TSC, from two familial and eight sporadic pedigrees), we screened mutations by single-strand conformation polymorphism and heteroduplex analysis (SSCP/HA) followed by direct sequencing. Disease causative mutations were detected in four patients. For the remaining six patients, we performed whole-coding exon sequencing of both genes, and mutations were identified in two more patients. For the remaining four patients, large deletion analysis (see below) was performed; one TSC1 and one TSC2 large deletion were detected. Details of these mutations have been reported previously.28
For the remaining 47 patients (including 37 definite and 10 suspect TSC, from 6 familial and 41 sporadic pedigrees), we applied CEL nuclease-mediated heteroduplex incision with polyacrylamide gel electrophoresis and silver staining (CHIPS) for mutation screening.35, 36 CHIPS is the simple and convenient system we have recently developed, possessing very high sensitivity for mutation screening as well or better than denaturing high-performance liquid chromatography (DHPLC). It is based on enzyme mismatch cleavage and optimizes each step of the system; that is, PCR protocol, heteroduplex formation, selection of endonuclease and DNA digesting condition and running gel matrix and developing methods. We reached 100% sensitivity and specificity by CHIPS in previous studies.35, 36 Hence, we did not perform full coding sequencing of TSC1/TSC2 for the patients whose mutations were not detected by CHIPS. PCR primer sets using this study for SSCP/HA or CHIPS were the same as described previously, for exons 1–41 of TSC215, 37 and exons 3–23 of TSC1.15
To detect large gene deletion or duplication of TSC1 or TSC2 in the all patients with no mutation detected by CHIPS screening, we performed long-range PCR as described by Dabora et al.,16 and Southern blotting followed by densitometry analysis28 for both genes.
Mutation nomenclature is in accordance with Dunnen et al.38 For TSC2, complementary DNA (cDNA) sequence is according to GenBank accession number NM_000548.3, transcript variant 1. This recent version of the cDNA sequence includes a 5′ leader exon as exon 1 and following 41 coding exons from exon 2–42. Although codon number was not changed, exon number was shifted from previously reported TSC2 mutations that do not count a leader exon. In this paper, we adhere to the previous exon numbering system of TSC2 for convenience in comparison. For TSC1, cDNA sequence is according to Genbank accession number NM_000368.4, transcript variant 1, including two 5′ leader exons (exon 1 and 2) and 22 coding exons (exon 3 to 23) without any changes from previous reports.
We reviewed all TSC mutations reported in Japanese patients available in the literature and compared them to TSC mutations reported in other ethnic populations. We found inconsistent mutation nomenclature in each paper and several errors in mutation descriptions in the reviewing process. We standardized and corrected all reported TSC mutations in Japanese patients and summarized them in a unified nomenclature system.38
Statistical analysis
Differences in each clinical trait were evaluated between TSC1 patients and TSC2 patients, TSC1 patients and no mutation identified (NMI) definite TSC patients and TSC2 patients and NMI-definite TSC patients. Fisher's exact test was used for the analysis of categorical variables, and P<0.05 (one-sided) was considered statistically significant. The quantitative variables—average of grade scales—were evaluated by F-test first and then equal variance data were analyzed by Student’s unpaired t-test, and unequal variance data were analyzed by Welch’s t-test. In both analyses P<0.05 (double-tailed) was considered statistically significant. To compare the incidences of TSC1 and TSC2 patients between Japanese studies and studies of other ethnic groups, χ2-test was used and P<0.05 was considered statistically significant.
Results
Identification and characterization of mutations
Among the total of 57 patients with TSC (46 definite and 11 suspect TSC), 11 TSC1 mutations (2 familial and 9 sporadic cases, all of them were definite TSC) and 20 TSC2 mutations (2 familial and 18 sporadic cases, including 19 definite and 1 suspect TSC) were detected. All TSC mutations detected by this study are summarized in Table 1. Overall mutation detection rate was 54.4% (31 among 57 patients) and 65.2% in definite TSC population (30 among 46 patients). Overall mutation detection rate by CHIPS was 48.9% (23 among 47 patients) and 62.2% in definite TSC (23 among 37 patients). Compared with the screening results of our first 10 patients (nine definite and one suspect TSC), this detection rate of CHIPS is reasonable and showing its high sensitivity. With the exclusion of two large gene deletions, six mutations were identified among the first 10 patients, of which, four mutations were positively screened by SSCP/HA analysis and remaining two were just detected by whole-coding exon sequencing of both TSC1 and TSC2.
The 11 TSC1 mutations included one missense, two splicing, two nonsense, five frameshift mutations and one large deletion. Eight of them are novel mutations detected by this study and two of them are previously reported only by us, whereas Q654X has been reported by the other US group.39 In TSC1, several amino acid substituted nucleic acid changes are found in general population and these amino acid substitutions are recognized as polymorphisms not as missense mutations. So, we evaluated one novel missense substitution H181P detected in this study for its pathogenesis. In the TSC1 mutation and polymorphism databases, H181P was not described in the human gene mutation database (http://www.hgmd.cf.ac.uk/ac/index.php), the Leiden open variation database (http://www.lovd.nl/2.0/index.php) or dbSNP (http://www.ncbi.nlm.nih.gov/snp/). So, four web-based algorithmic programs were applied to predict the effect of amino acid substitution on protein function. PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/),40 PMut (http://mmb2.pcb.ub.es:8080/PMut/)41 and Mutation taster (http://www.mutationtaster.org/index.html)42 predicted that this substitution is pathological and SIFT (http://sift.jcvi.org/)43 predicted that it is tolerated with relatively low credibility. H181P did not appear in other TSC patients we analyzed in this study nor in the DNA of the 50 normal Japanese volunteers. According to these results, we consider that H181P is a disease causative mutation.
Another TSC1 missense substitution, T899S, appeared in family 15 who were diagnosed as suspect TSC with the only presenting TSC phenotype was hypomelanotic macules of the skin. The same substitution was previously reported as a causative mutation in a Japanese patient with TSC.29 T899S was in agreement with the record of dbSNP rs76801599, in which clinical significance is not tested. Although T899S does not appear in the other TSC patients in this study, nor in the 50 Japanese normal volunteers; all of the four web-based programs mentioned above predicted that this substitution does not affect protein function. According to these results, we conclude that T899S is a rare polymorphism in the Japanese population. In family 10, TSC1 large deletion was detected by long-range PCR, which spans exon 17–19 with somatic mosaicism of two different deletions. The details of this patient have been reported previously.28
The 20 TSC2 mutations included four missense, four in-frame deletion, one stop codon, four splicing, six frameshift mutations and one large deletion were detected in our patients. However, no TSC2 nonsense mutation was detected. Seven of them are novel mutations detected in this study and two of them are previously reported only by us, whereas the other 11 have been reported previously in other ethnic groups.16, 17, 19, 37, 44, 45, 46 The stop codon mutation detected in family 31 is the same mutation as previously reported.46 TG deletion of the stop codon (TGA) causes continuation of translation beyond the stop codon, without a new stop codon downstream. Goedbold et al.46 demonstrated that this mutated tuberin could not bind to hamartin by the yeast two-hybrid experiment. The TSC2 patient of family 1 had only a SEGA as a feature of TSC and was classified as suspect TSC by clinical diagnostic criteria. This patient was included in our first 10 patients and mutational screening was performed by SSCP/HA with SEGA-derived DNA at the first instance. After detected TSC2 missense mutation R1743Q in the brain tumor, we screened whether the same mutation existed in other tissues of the patient including blood, hair, nail, buccal mucosa and urinary sediment, but R1743Q was not detected in these tissues. We concluded that R1743Q was a somatic mutation in SEGA and TSC2 LOH was also confirmed in this tumor. Details of this TSC2 somatic mosaic patient have been described elsewhere.47
Analysis of clinical manifestations and genotype–phenotype correlation
The clinical features associated with each TSC1 or TSC2 mutation-positive patients and patients with NMI are displayed in Table 2. A comparison of the incidence and grade score of these clinical manifestations between the TSC1, TSC2 and NMI-definite TSC patients are summarized in Table 3.
Among the clinical findings, subependymal nodule and cortical tuber are the most consistent features in all groups, and at least one of these features were found in all patients with TSC1 and TSC2 patients who were given brain imaging tests. However, two patients with NMI-definite TSC (family 34 and 57) had no brain lesions. Among the neurological findings, incidence of any developmental delay or mental retardation (grade 1 or 2) was significantly higher in TSC2 patients (12/15) in comparison with TSC1 patients (4/11; P=0.032, Fisher's exact test). Also, the incidence of substantial developmental delay or mental retardation (grade 2) was significantly higher in TSC2 patients (9/15) in comparison with TSC1 patients (2/11; P=0.040, Fisher's exact test). In addition, mean grade score was significantly higher in TSC2 patients (1.4) than TSC1 patients (0.5; P=0.015, Student’s unpaired t-test). This trend of worse psychomotor delay in TSC2 patients was also observed between TSC2 patients and NMI-definite TSC patients, in incidence of grade 1 or 2 (6/16 vs 12/15, P=0.020, Fisher’s exact test), grade 2 (3/16 vs 9/15, P=0.023, Fisher's exact test) and mean grade score (0.6 vs 1.4, P=0.005, Student’s unpaired t-test). However, no difference was observed between TSC1 patients and NMI-definite TSC patients. The incidence and severity of epilepsy, brain imaging and retinal hamartoma were not different in TSC1, TSC2 and NMI-definite TSC patients.
From the phenotypic skin lesions, only ungula fibroma was observed more in TSC1 patients than in TSC2 patients (P=0.034, Fisher's exact test) but no difference was detected in facial angiofibroma, hypomelanotic macules and other skin lesions. Renal, hepatic, pulmonary and cardiac lesions were variable in all groups and the incidences and mean grade scores were not statistically different. We also examined the data for genotype/phenotype correlation and found no correlation between mutation type and phenotype in both TSC1 and TSC2 patients.
Mutation distributions of TSC1 and TSC2 genes in this study, as well as those reported in Japanese patients and previous studies of other races are summarized in Table 4. No obvious difference was indicated between the Japanese population and other ethnic groups, except for low frequencies of TSC2 nonsense mutation (P=0.015, this study vs US and Europe studies; and P=0.000, all reported Japanese mutations vs US and Europe studies, χ2-test). Incidence of TSC1 and TSC2 mutation among patients with TSC are compared with either this study or the sum of Japanese cohort studies and other previous studies from different ethnic groups (Table 5). It is remarkable that incidence of TSC1 patients in Japan is significantly higher than in other studies. To compare US with Europe studies, overall TSC2/TSC1 detected mutation ratio is lower in this study (1.7 vs 3.7, P=0.043, χ2-test) and in the Japanese cohort (1.9 vs 3.7, P=0.014, χ2-test). When comparing only sporadic cases, these variances are much more significant, both in this study (1.9 vs 5.5, P=0.009, χ2-test) and in the Japanese cohort (2.2 vs 5.5, P=0.007, χ2-test).
Discussion
We screened both TSC1 and TSC2 for disease causative mutations in 57 Japanese families with TSC, including 46 definite and 11 suspect TSC, and identified 11 TSC1 mutations and 20 TSC2 mutations including one somatic mosaic mutation in the patient with suspect TSC (family 1). Mutation detection rate in definite TSC patients was 30/46 (65.2%). This rate is comparable to previous large cohort studies. In addition, no mutation was detected in suspect TSC patients other than the TSC2 somatic mosaic case mentioned above.
First, this result proved the accuracy and utility of TSC clinical diagnostic criteria in general,3 but a small fraction of suspect TSC could have TSC mutation. Au et al.17 also showed similar results in large-scale cohort. They screened TSC mutations in 368 patient with TSC (including 325 definite TSC and 43 uncertain diagnosis or not-definite TSC patients), and disease causative mutations were detected in a total of 243 patients, of which, 234 were definite TSC, eight were patients with uncertain diagnosis because of poor medical records and one TSC2 mutation was found in a not-definite TSC patient.
We could not identify TSC1 or TSC2 mutations in 16 definite TSC patients. Two of them have no brain lesions and no neurological symptoms (epilepsy, mental retardation, subependymal nodule, SEGA or cortical tuber). A 25-year-old male (family 57) has facial angiofibroma and renal AML, but no other TSC-related lesions. A 70-year-old female (family 34) has hypomelanotic macules, ungual fibroma, renal AML and pulmonary LAM, but no facial angiofibroma. As represented in these patients, several NMI-definite TSC patients seem to abortive type although fulfilled clinical diagnostic criteria.
It is reported that a significant population up to 10% of TSC patients have somatic mosaicism and it might be difficult to detect the mutations by peripheral blood DNA testing for these patients.15, 16 Actually, in family 1, we detected a TSC2 somatic mosaic mutation only in the tumor DNA sample. If we could test tumor samples of these NMI-definite TSC families, it may increase the possibility to detect more disease causative TSC mutations. From another viewpoint, we might have missed TSC1 or TSC2 mutations in these patients due to technical problems. For point mutations, or small insertions or deletions, we detected a sufficient number of mutations; considering the high sensitivity of CHIPS technology and comparing it with the previous study using DHPLC,22 it is less possible that we missed many small mutations in this population. For large deletions, we checked large intragenic rearrangements by long-range PCR and large gene deletions including the 5′ or 3′ end of the genes by Southern blot analysis. Although we did not perform multiplex ligation-dependent probe amplification (MLPA) for each exon, previous studies indicate that point exon deletion is a rare event for both TSC1 and TSC2,19 and the majority of TSC2 large deletions could be detected by our methods. In addition, in our 16 NMI-definite TSC patients, nine patients have TSC2 polymorphisms and seven patients have TSC1 polymorphisms. Existence of polymorphisms proved two copies of each gene at these sites, and previous studies indicate that TSC2 large deletions are detected in only 8% of TSC2 patients at the maximum.16 These data indicate it is less possible that we missed many large TSC1 or TSC2 deletions. Also, previous linkage analysis confirmed that there is no other TSC locus.
Another possibility could be hidden in the large 3′-UTR sequence in TSC1 gene that has not yet been analyzed by us or any other researchers. TSC1 cDNA consists of 234-bp 5′-UTR, 3045-bp coding and 5097-bp 3′-UTR sequence. Although many polymorphisms are reported in dbSNP, no pathological mutation has been detected in TSC1 3′-UTR, and the function of this long 3′-UTR sequence is still unknown. Put together with NMI-definite TSC patients that resemble TSC1 patients in that they manifest intellectually milder phenotype than TSC2 patients, precious analysis of this sequence is an issue in the future for determining the possibility that we missed TSC1 3′-UTR mutations in NMI-definite TSC population.
The spectrum and distribution of TSC1 and TSC2 mutations in this study were not different from previous reports, except for the low TSC2 nonsense mutation finding. To date, including this study, 92 mutations (33 TSC1, 59 TSC2) of Japanese patients with TSC are reported in the literature, and no ethnically specific mutational hot spots are detected (Figure 1, Supplementary Table 1). The only reported TSC2 nonsense mutation in Japanese patients is R1459X in exon 33 found in two different families.29 Why the TSC2 nonsense mutation is rare in the Japanese TSC population is not clear at this time. We have to collect and analyze more TSC mutations in Japanese patients to elucidate the reason.

Summary of TSC mutations reported in Japanese patients. Mutations reported in this study are indicated in the upper portion and mutations reported by others are indicated in the lower portion of each gene pattern diagram (a, Zhang et al.;29 b, Yamashita et al.;30 c, Yamamoto et al.;31 d, Sasongoko et al.32 and e, Sato et al.33). Missense mutations and in-frame deletions are described by amino acid changes. Δ, frameshift mutation; ↑, splicing mutation; × , nonsense mutation; ◊, stop codon mutation; x2, reported two other non-related families.
Comparing the phenotypes of TSC1, TSC2 and NMI-definite TSC patients, we conclude that TSC2 patients have much profound developmental delay or mental retardation than TSC1 patients and NMI-definite TSC patients. It is difficult to evaluate mental retardation of very young patients with the same scale because the patients were collected from different hospitals all over Japan. However, in clinical practice, evaluating developmental delay by physical examination and developmental scale is routine work for pediatric neurologists. So, our modified check list precisely evaluates developmental state even in young patients and also evaluates mental retardation of older patients per the original check list item.16 The milder phenotypes of TSC1 patients16, 17 and NMI patients17 in comparison with TSC2 patients have also been reported elsewhere. Our data indicate that this is consistent in the Japanese population. However, physicians must be cautious in two issues. First, a milder phenotype just means developmental delay or mental retardation, and there are no differences of incidence or severity in all other findings between TSC1 and TSC2 patients including skin, renal, cardiac, pulmonary manifestations and even epilepsy and brain images. Although previous studies have indicated a milder renal phenotype16, 17 and facial angiofibroma17 in TSC1 patients, we could not detect these differences because of low patient numbers and actual phenotypic differences are slight. Second, even though overall incidence and severity of intellectual disability are milder in TSC1 than TSC2, some TSC1 patients have profound developmental delay or mental retardation as found in TSC2 patients. These points should be kept in mind, especially when providing genetic counseling.
We also identified a higher incidence of ungual fibroma in TSC1 patients than in TSC2 patients. However, ungual fibroma is a lesion that appears in adults, especially after middle age, the results was affected by younger TSC2 patients ages in our cohort (average 10.2 years old, one 21-year-old patient has ungula fibroma) than TSC1 patients (average 26.5 years old, four 50, 48, 22, 15-year-old patients have ungual fibroma).
One notable difference in the Japanese TSC population in comparison with previous studies in US and Europe is the higher TSC1 patient incidence among all mutation-positive TSC patients. The ratio of TSC2/TSC1 mutation-positive cases is 1.7 in this study and is 1.9 after adding two other Japanese cohort studies, compared with 3.4–5.6 in US and Europe studies (3.7 as a whole). Though TSC1 patients are more detected in familial cases than in sporadic cases—because of higher reproductive fitness by its milder phenotype of intellectual faculties—higher incidence of TSC1 patients in Japan is not dependent on a high familial case ratio of mutation screened. Rather, in this study, familial cases comprise only 8 families among 46 definite TSC, and a higher TSC1 patient ratio is more evident when comparing only the sporadic cases: 1.9 in this study and 2.2 in Japanese cohort to 4.3–8.5 in US and Europe (5.5 as a whole). Other than US and Europe, TSC2/TSC1 mutation ratio is 6.1 in Taiwan and 2.7 in Australia. In Korea, one TSC mutation study showed a high incidence of TSC1 patients (TSC2/TSC1 is 1.3)24 but another study did not (5.5).23 We cannot conclude TSC1 incidence in Korea because overall mutation detection ratios in both of these studies are low. In any case, it seems that incidence of TSC1 patients among TSC differs across race and region.
In this study, eight TSC1 mutations are novel and many other TSC1 mutations detected in Japanese patients in other studies are also novel and unique. By contrast, many TSC2 mutations detected in the Japanese population are also reported in US and Europe studies. It seems that the Japanese population has a specific TSC1 mutational variation.
The reason why TSC1 patients are detected at a higher incidence in the Japanese TSC population is not clear. As mentioned above, it is not likely that we missed many TSC2 mutations. TSC1 mutations detected in the Japanese population are scattered throughout TSC1 without hot spots. A majority of TSC1 mutations in Japanese patients are detected in sporadic cases and TSC1 familial clusters are unlikely to exist. It is difficult to imagine that mutation ratio of the specific gene in the specific race or region would increase without the founder effect.
Another point of view is a social environmental factor. Differences of the medical service system or health disparity, including healthcare insurance and adoption rate of computed tomography and magnetic resonance units in the regions, may affect the results. TSC1 patients could be more frequently diagnosed in Japan owing to a generous healthcare insurance system and the convenient medical situation. Because TSC1 patients manifest a milder phenotype than TSC2 patients, it could be that less TSC1 patients visit a hospital in other countries, which does not have universal healthcare system like the United States. In addition, the adoption rate of computed tomography/magnetic resonance units in clinics and hospitals in Japan is world’s best.48 It seems that brain and abdominal imaging studies are performed more frequently in Japan compared with other countries including US and Europe, which consequently increases the chance for diagnosis of TSC among patients with milder phenotype. If this is true, many TSC1 patients are still undiagnosed in other countries. More research and analysis for TSC1 mutations are vitally needed.
Accession codes
References
Gomez, M. R., Sampson, J. R. & Whittemore, V. H. Tuberous Sclerosis Complex 3rd edn Oxford University Press: Oxford, (1999).
Bourneville, D. M. Sclerose tubereuse des circonvolutions cerebrales: idiotie et epilepsie hemiplegique. Arch. Neurol. 1, 81–91 (1880).
Roach, E. S. & Sparagana, S. P. Diagnosis of Tuberous sclerosis complex. J. Child. Neurol. 19, 643–649 (2004).
Kwiatkowski, D. J. & Short, M. P. Tuberous sclerosis. Arch. Dermatol. 130, 348–354 (1994).
Osborne, J. P., Fryer, A. & Webb, D. Epidemiology of tuberous sclerosis. Ann. N. Y. Acad. Sci. 615, 125–127 (1991).
van Slegtenhorst, M., de Hoogt, R., Hermans, C., Nellist, M., Janssen, B., Verhoef, S. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808 (1997).
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993).
Jozwiak, J., Jozwiak, S. & Wlodarski, P. Possible mechanisms of disease development in tuberous sclerosis. Lancet. Oncol. 9, 73–79 (2008).
Kwiatkowski, D. J. Rhebbing up mTOR: new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer. Biol. Ther. 2, 471–476 (2003).
Niida, Y., Stemmer-Rachamimov, A. O., Logrip, M., Tapon, D., Perez., R., Kwiatkowski, D. J. et al. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am. J. Hum. Genet. 69, 493–503 (2001).
Orlova, K. A. & Crino, P. B. The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 1184, 87–105 (2010).
Han, S., Santos, T. M., Puga, A., Roy, J., Thiele, E. A., McCollin, M. et al. Phosphorylation of tuberin as a novel mechanism for somatic inactivation of the tuberous sclerosis complex proteins in brain lesions. Cancer. Res. 64, 812–816 (2004).
Ess, K. C. Tuberous sclerosis complex: a brave new world? Curr. Opin. Neurol. 23, 189–193 (2010).
Rosner, M., Hanneder, M., Siegel, N., Valli, A. & Hengstschläger, M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 658, 234–246 (2008).
Niida, Y., Lawrence-Smith, N., Banwell, A., Hammer, E., Lewis, J., Beauchamp, R. L. et al. Analysis of both TSC1 and TSC2 for germline mutations in 126 unrelated patients with tuberous sclerosis. Hum. Mutat. 14, 412–422 (1999).
Dabora, S. L., Jozwiak, S., Franz, D. N., Roberts, P. S., Nieto, A., Chung, J. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68, 64–80 (2001).
Au, K. S., Williams, A. T., Roach, E. S., Batchelor, L., Sparagana, S. P., Delgado, M. R. et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet. Med. 9, 88–100 (2007).
Jones, A. C., Shyamsundar, M. M., Thomas, M. W., Maynard, J., Idziaszczyk, S., Tomkins, S. et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 64, 1305–1315 (1999).
Rendtorff, N. D., Bjerregaard, B., Frödin, M., Kjaergaard, S., Hove, H., Skovby, F. et al. Analysis of 65 tuberous sclerosis complex (TSC) patients by TSC2 DGGE, TSC1/TSC2 MLPA, and TSC1 long-range PCR sequencing, and report of 28 novel mutations. Hum. Mutat. 26, 374–383 (2005).
Sancak, O., Nellist, M., Goedbloed, M., Elfferich, P., Wouters, C., Maat-Kievit, A. et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur. J. Hum. Genet. 13, 731–741 (2005).
Chopra, M., Lawson, J. A., Wilson, M., Kennedy, S. E., Taylor, P., Buckley, M. F. et al. An Australian tuberous sclerosis cohort: are surveillance guidelines being met? J. Paediatr. Child. Health. 47, 711–716 (2011).
Hung, C. C., Su, Y. N., Chien, S. C., Liou, H. H., Chen, C. C., Chen, P. C. et al. Molecular and clinical analyses of 84 patients with tuberous sclerosis complex. BMC Med. Genet. 7, 72 (2006).
Choi, J. E., Chae, J. H., Hwang, Y. S. & Kim, K. J. Mutational analysis of TSC1 and TSC2 in Korean patients with tuberous sclerosis complex. Brain. Dev. 28, 440–446 (2006).
Jang, M. A., Hong, S. B., Lee, J. H., Lee, M. H., Chung, M. P., Shin, H. J. et al. Identification of TSC1 and TSC2 mutations in Korean patients with tuberous sclerosis complex. Pediatr. Neurol. 46, 222–224 (2012).
Khare, L., Strizheva, G. D., Bailey, J. N., Au, K. S., Northrup, H., Smith, M. et al. A novel missense mutation in the GTPase activating protein homology region of TSC2 in two large families with tuberous sclerosis complex. J. Med. Genet. 38, 347–349 (2001).
Jansen, A. C., Sancak, O., D'Agostino, M. D., Badhwar, A., Roberts, P., Gobbi, G. et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann. Neurol. 60, 528–539 (2006).
Brook-Carter, P. T., Peral, B., Ward, C. J., Thompson, P., Hughes, J., Maheshwar, M. M. et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome. Nat. Genet. 8, 328–332 (1994).
Wakisaka, A. DNA diagnosis of ten Japanese patients with tuberous sclerosis complex: Development of systemic mutational analysis of TSC genes and and certification of sensitivity for clinical application. Juzen. Med. Soc. 115, 62–74 (2006).
Zhang, H., Nanba, E., Yamamoto, T., Ninomiya, H., Ohno, K., Mizuguchi, M. et al. Mutational analysis of TSC1 and TSC2 genes in Japanese patients with tuberous sclerosis complex. J. Hum. Genet. 44, 391–396 (1999).
Yamashita, Y., Ono, J., Okada, S., Wataya-Kaneda, M., Yoshikawa, K., Nishizawa, M. et al. Analysis of all exons of TSC1 and TSC2 genes for germline mutations in Japanese patients with tuberous sclerosis: report of 10 mutations. Am. J. Med. Genet. 90, 123–126 (2000).
Yamamoto, T., Pipo, J. R., Feng, J. H., Takeda, H., Nanba, E., Ninomiya, H. et al. Novel TSC1 and TSC2 mutations in Japanese patients with tuberous sclerosis complex. Brain. Dev. 24, 227–230 (2002).
Sasongko, T. H., Wataya-Kaneda, M., Koterazawa, K., Gunadi, Yusoff, S., Harahap, I. S. et al. Novel mutations in 21 patients with tuberous sclerosis complex and variation of tandem splice-acceptor sites in TSC1 exon 14. Kobe. J. Med. Sci. 54, E73–E81 (2008).
Sato, T., Seyama, K., Fujii, H., Maruyama, H., Setoguchi, Y., Iwakami, S. et al. Mutation analysis of the TSC1 and TSC2 genes in Japanese patients with pulmonary lymphangioleiomyomatosis. J. Hum. Genet. 47, 20–28 (2002).
Lahiri, D. K. & Schnabel, B. DNA isolation by a rapid method from human blood samples: effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem. Genet 31, 321–328 (1993).
Tsuji, T. & Niida, Y. Development of a simple and highly sensitive mutation screening system by enzyme mismatch cleavage with optimized conditions for standard laboratories. Electrophoresis 29, 1473–1483 (2008).
Niida, Y., Kuroda, M., Mitani, Y., Okumura, A. & Yokoi, A. Applying and testing the conveniently optimized enzyme mismatch cleavage method to clinical DNA diagnosis. Mol. Genet. Metab. 107, 580–585 (2012).
Beauchamp, R. L., Banwell, A., McNamara, P., Jacobsen, M., Higgins, E., Northrup, H. et al. Exon scanning of the entire TSC2 gene for germline mutations in 40 unrelated patients with tuberous sclerosis. Hum. Mutat. 12, 408–416 (1998).
den Dunnen, J. T. & Antonarakis, S. E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12 (2000).
Kwiatkowska, J., Jozwiak, S., Hall, F., Henske, E. P., Haines, J. L., McNamara, P. et al. Comprehensive mutational analysis of the TSC1 gene: observations on frequency of mutation, associated features, and nonpenetrance. Ann. Hum. Genet. 62, 277–285 (1998).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods. 7, 248–249 (2010).
Ferrer-Costa, C., Gelpí, J. L., Zamakola, L., Parraga, I., de la Cruz, X. & Orozco, M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21, 3176–3178 (2005).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 7, 575–576 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Jones, A. C., Sampson, J. R., Hoogendoorn, B., Cohen, D. & Cheadle, J. P. Application and evaluation of denaturing HPLC for molecular genetic analysis in tuberous sclerosis. Hum. Genet. 106, 663–668 (2000).
Gilbert, J. R., Guy, V., Kumar, A., Wolpert, C., Kandt, R., Aylesworth, A. et al. Mutation and polymorphism analysis in the tuberous sclerosis 2 (TSC2) gene. Neurogenetics 1, 267–272 (1998).
Goedbloed, M. A., Nellist, M., Verhaaf, B., Reuser, A. J., Lindhout, D., Sunde, L. et al. Analysis of TSC2 stop codon variants found in tuberous sclerosis patients. Eur. J. Hum. Genet. 9, 823–828 (2001).
Ichikawa, T., Wakisaka, A., Daido, S., Takao, S., Tamiya, T., Date, I. et al. A case of solitary subependymal giant cell astrocytoma: two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J. Mol. Diagn. 7, 544–549 (2005).
Nakajima, Y., Yamada, K., Imamura, K. & Kobayashi, K. Radiologist supply and workload: international comparison--Working Group of Japanese College of Radiology. Radiat. Med. 26, 455–465 (2008).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a grant from the Ministry of Health, Labor and Welfare of Japan, Tokyo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Niida, Y., Wakisaka, A., Tsuji, T. et al. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J Hum Genet 58, 216–225 (2013). https://doi.org/10.1038/jhg.2013.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.3
Keywords
This article is cited by
-
Renal phenotypes correlate with genotypes in unrelated individuals with tuberous sclerosis complex in China
Orphanet Journal of Rare Diseases (2022)
-
TSC2 somatic mosaic mutation, including extra-tumor tissue, may be the developmental cause of solitary subependymal giant cell astrocytoma
Child's Nervous System (2022)
-
Mutational analysis of renal angiomyolipoma associated with tuberous sclerosis complex and the outcome of short-term everolimus therapy
Scientific Reports (2019)
-
Pattern of TSC1 and TSC2 germline mutations in Russian patients with tuberous sclerosis
Journal of Human Genetics (2018)
-
Establishment of a Regional Interdisciplinary Medical System for Managing Patients with Tuberous Sclerosis Complex (TSC)
Scientific Reports (2018)
