
Abstract
Pseudomyxoma peritonei (PMP) is a rare abdominal malignancy. We hypothesized that next-generation exomic sequencing would identify recurrent mutations that may have prognostic or therapeutic implications. Ten patients were selected on the basis of availability of tissue and adequate follow-up. They were treated at our institution between September 2002 and August 2004. Using next-generation exomic sequencing, we tested for mutations in 236 cancer-related genes in formalin-fixed paraffin-embedded slides. MCL1 amplification was additionally tested with immunohistochemical staining. Detectable mutations were found in 8 patients (80%). Seven patients harbored a KRAS mutation, most commonly involving codon 12. Four GNAS mutations (R201H/R201C substitutions) were also detected. MCL1 and JUN were concurrently amplified in three patients. One patient with MCL1 and JUN amplification had concurrent amplification of MYC and NFKBIA. ZNF703 was amplified in one patient. Patients with MCL1 amplification were also found to express MCL1 with immunohistochemistry, but MCL1 expression was also detected in some patients without amplification. To our knowledge, we are the first to report MCL1 and JUN coamplification in PMP. Expression of MCL1 may not be completely dependent on amplification. The prognostic and therapeutic implications of these recurrent mutational events are the subject of ongoing investigation.
Similar content being viewed by others

Introduction
Pseudomyxoma peritonei (PMP) is a rare gastrointestinal malignancy1 that is characterized by low-grade mucin-producing metastatic implants on peritoneal surfaces. Although patients with PMP may have an indolent course, they frequently experience recurrent accumulations of gelatinous mucin. The natural history and biology of PMP are distinctive from colorectal carcinoma, and PMP is predominantly locoregionally aggressive. The primary treatment for PMP is surgical debulking,2 and when there is a recurrence, further cytoreductive surgery and hyperthermic intraperitoneal (IP) chemotherapy can be considered.3 One center reported a median survival of 6.72 years in patients treated with aggressive cytoreductive surgery and hyperthermic IP chemoperfusion.4 Our center uses IP chromic phosphate P32 and fluorouracil after maximal surgical resection,5 and the overall survival with this approach was 8.1 years. Patients who had optimal surgical cytoreduction received postoperative IP chemotherapy and experienced a median survival of 23.5 years, whereas patients who did not achieve optimal surgical cytoreduction and who did not receive postoperative IP chemotherapy had a median survival of 7.5 years.5
Because PMP is encountered infrequently, few studies have been done on the molecular alterations of this malignancy. Many of the studies to date have focused on mutations commonly seen in colorectal cancer, particularly KRAS and its prognostic importance.6, 7, 8 In this study, samples from 10 patients with PMP were tested for the presence of a genetic mutation by next-generation sequencing technology. The genomic changes were comprehensively profiled on an individual basis. We hypothesized that next-generation exomic sequence analysis would lead to the discovery of recurrent and common mutational markers in PMP, which may lead to the identification of potential molecular targets for personalized oncological therapy.
Materials and methods
Patient selection and treatment
The study was approved by the Mayo Clinic Institutional Review Board. Ten patients with a diagnosis of PMP were selected on the basis of availability of formalin-fixed paraffin-embedded (FFPE) archival slides, adequacy of clinical notes and duration of follow-up. These patients were treated at Mayo Clinic, Rochester, MN, USA between September 2002 and August 2004. A retrospective medical record review of these 10 patients was completed in March 2013. All patients underwent surgical resection, with or without postoperative chemotherapy. The surgical intent was definitive for all patients, and operative reports were reviewed. Intraoperatively, all patients had bulky and peritoneally disseminated disease, and all of their submitted surgical specimens were stored and available for analysis. Patients whose disease was debulked to a residual disease no greater than 3 mm were considered optimally debulked and received IP chemotherapy postoperatively. The postoperative regimen has been described previously in detail,5 namely, IP fluorouracil and chromic phosphate P32, a β-emitting colloid with an activity of 10 mCi. Fluorouracil was given daily for 3 consecutive days, followed by IP phosphate P32, which was given the day after the IP fluorouracil infusions were complete.
Mutational analyses and detection
Original hematoxylin and eosin slides were reviewed for the presence of invasive cystadenocarcinoma or adenocarcinoma cells by one of us, a surgical pathologist (RPG) to confirm the pathological diagnosis of PMP. The presence of an invasive component by PMP was confirmed in all 10 cases, which were used in the genome-wide analysis. The clinicopathological Ronnett’s criteria9 were applied retrospectively. The sections with the highest ratio of tumor to nonmalignant tissues were selected. No control was used in this study. Archival FFPE slides were then obtained and cut into 10 separate 5-μm sections. Mutations in DNA in 236 cancer-related genes, which included at least 3230 exons and 47 introns from 19 genes often rearranged or altered in somatic human cancer, were then tested using the Illumina HiSeq2000 platform (Foundation Medicine, Cambridge, MA, USA) (Supplementary Table 1). Next-generation sequencing technology is capable of genotyping individual base pairs by detecting multiple copies of exonal mutations and fusion genes. For our assay, the power of average coverage for uniquely mapping reads is > × 900.10 Sequence data are used for a proprietary data analysis by Foundation Medicine, as described elsewhere,11 that includes mapping to a reference human genome and comparing genomic DNA sequence coverage with a process-matched normal control sample. Additionally, we performed a post hoc immunohistochemical analysis (Histoserv, Inc., Germantown, MD, USA), using MCL1 (myeloid cell leukemia sequence 1) primary antibody staining on FFPE slides that were sectioned into 3-μm slides and stained with MCL1 monoclonal antibody (ab31948; Abcam, Cambridge, MA, USA).
Statistical methods
Descriptive statistics were used to summarize patient, tumor and mutational characteristics and were reported as median and range or number and frequency. The χ2 testing (new reference) was performed for trend estimates and correlation of mutational status in patient samples.12 The χ2 analysis examined the distribution of attributes between the two sets of observations and performed a goodness-of-fit evaluation of the samples. P value of ⩽0.05 was considered statistically significant. Survival since diagnosis was calculated according to the log-rank Kaplan–Meier method.13 All tests were two sided, with 5% type I error rates.
Results
Clinical and tumor characteristics
Selected patient and tumor characteristics are shown in Table 1. All patients had PMP evident at the time of surgery as low-grade mucinous neoplasms of the appendix with abundant intra-abdominal mucin and scant neoplastic cells. Eight of the 10 cases were low grade, with 2 cases (patients 1 and 10) with moderate cytological atypia noted as grade 3 (of 4 maximum) at our institution. None of these cases was high grade (that is, severe atypia, grade 4 of 4). The median follow-up was 5.5 years (range, 2.0–9.3 years) for all 10 patients, and 8.0 years (range, 4.3–9.3 years) in 4 patients who remained alive at the time of this analysis. The most common presentation was abdominal pain, which occurred in seven patients, with a median duration of 21 days (range, 0–135 days) between the onset of symptoms and evaluation by a medical provider. Additionally, patients also experienced weight loss (in four) and fatigue (in three) but rarely fever (in one) or bowel obstruction (none). On physical examination, ascites was a common finding, occurring in seven patients but rarely found were a palpable mass (in one) or peritoneal signs (in two). All patients underwent exploratory laparotomy during which the analyzed tissue samples were obtained, and 9 of the 10 patients had >1 organ resected in addition to the peritoneum, appendix and colon (most commonly, omentum (8 of the 9 patients); 3 had splenectomy as well). Six patients received IP fluorouracil for 3 days followed by IP phosphorus P32.
MCL1 and JUN amplifications
Eight of the 10 samples had ⩾1 detectable mutation (Table 2). The median number of genetic alterations was 2 (range, 0–4). Two samples had no identifiable mutations. MCL1 and JUN were concurrently amplified in three samples, of which two had concurrent KRAS codon 12 and GNAS codon 201 substitutions. One sample with MCL1 and JUN amplification had concurrent amplification of MYC and NFKBIA.
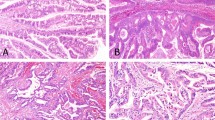
Additional slides were prepared for immunohistochemical staining for cytoplasmic expression of MCL1 (Figure 1). Three samples had moderate-to-strong staining, five had moderate staining and two had weak staining. MCL1 expression was detected in samples with MCL1 amplification; however, MCL1 expression was also detected in other samples that did not have MCL1 amplification.

Immunohistochemical staining for MCL1 in two patient samples demonstrated moderate (left, patient 2) and moderate-to-strong (right, patient 6) cytoplasmic staining. Both patients had unique MCL1 and JUN coamplification in samples of pseudomyxoma peritonei tested by next-generation genetic sequencing (original magnification × 200, left; × 100, right).
KRAS, GNAS and other mutations
The specific KRAS and GNAS mutations for each patient are summarized in Table 3. KRAS mutations were detected in seven patients. Six of the seven KRAS mutations involved codon 12, and one involved codon 59. Aspartic acid replaced glycine in four of these mutations (G12D). Valine (G12V) and alanine (G12A) each replaced glycine in the other two codon 12 mutations. We also detected a single KRAS A59T mutation. We detected four GNAS mutations, which manifested as two R201H and two R201C substitutions each. All four of these samples also had concurrent KRAS codon 12 substitutions. ZNF703 was amplified in one sample with a KRAS G12A substitution, without an associated GNAS mutation. Comutations in both codons 12 and 59 of KRAS were not found.
Survival
At the time of analysis, 6 patients (60%) had died. The median overall survival for this group of 10 patients was 5.3 years (Figure 2). Of the 10 patients, 9 experienced recurrence, including 7 locoregionally and 2 with persistent disease. Five patients underwent repeat attempts at cytoreduction for symptomatic disease recurrence (two with additional organs removed). Salvage chemotherapy regimens included two cycles of irinotecan, fluorouracil and leucovorin, then two cycles of irinotecan and cetuximab (in one patient) and leucovorin, fluorouracil and oxaliplatin (in two patients).

Overall survival of 10 patients with pseudomyxoma peritonei.
The coamplification of MCL1 or JUN demonstrated a slight trend favoring prolonged survival (median not reached vs 4.8 years, χ2=2.93, Wilcoxon P=0.09). Four patients were alive at the time of analysis; all three patients with both MCL1 and JUN amplified were still alive at 7.3, 4.3 and 8.7 years (patients 3, 8 and 9, respectively). Patient 3 was the only patient living without evidence of disease. Patient 6, alive and with the longest follow-up of 9.3 years, did not have MCL1 and JUN coamplification.
Discussion
We found mutations in 80% of our patients with PMP. We are the first, to our knowledge, to report coamplification of MCL1 and JUN in PMP, and we confirm previous findings of KRAS and GNAS mutations in PMP.
MCL1 is a BCL2-related anti-apoptotic gene important for myeloid differentiation.14 It has been mapped to the long arm of the first chromosome (1q21), an area that is rearranged in many malignancies.15 MCL1 is upregulated in chronic lymphocytic leukemia,16 hepatocellular carcinoma,17 breast cancer18 and non-small cell lung cancer.19 Overexpression of MCL1 in breast cancer was correlated with higher grade and poor survival.18 In at least one patient in the current study, amplification of MCL1 was associated with amplification of MYC. Coexpression of MCL1 and MYC is associated with poor survival in non-small cell lung cancer.20 MCL1 can potentially be targeted.21 MCL1 overexpression appears to have a key role in the resistance of some cancers to chemotherapy, particularly fluorouracil, which is often given IP for PMP. Because of MCL1’s short half-life, inhibiting its expression or neutralizing its function can induce rapid apoptosis in several cancer cell lines.22
JUN is a proto-oncogene similar to the transforming gene of avian sarcoma virus 17.23 This gene has no introns and is mapped to the short arm of chromosome 1 at 1p32-p31.24, 25 JUN is commonly expressed in gastroenteropancreatic neuroendocrine tumors,26 and its expression is correlated with worse survival in squamous cell lung cancers.27
KRAS mutations have been reported in mucinous appendiceal adenocarcinomas. One recent study identified KRAS mutations in 37 of 64 patients (57.8%).28 Of the 35 patients with specific mutation data, 31 had a codon 12 mutation, and the other 4 had a codon 13 mutation. There was no difference in the distribution of KRAS mutations between low-grade and high-grade PMPs, nor was there a difference in survival between those with or without a KRAS mutation. Similarly, another earlier study identified KRAS mutations in 8 of 16 samples (50%).29 The most common mutation was KRAS G12A, but G12T and G13A substitutions were also identified. More recently, a slightly higher rate of 61.3% of KRAS mutations in PMP of the appendix was reported.4 Previous studies have also reported a substantial incidence of KRAS mutations in their PMP patients, which are similar to our findings.30, 31 Although KRAS was not prognostic in our study or in previous studies of PMP, it has been associated with poor prognosis in colorectal cancer.32, 33, 34
GNAS R201H is an activating mutation of the α-subunit of a G-protein that is coupled to seven transmembrane receptors, leading to constitutive activation of adenylyl cyclase.35, 36 More recent work identified GNAS R201H substitution in half the low-grade appendiceal mucinous neoplasms that they studied but not in any mucinous adenocarcinomas.37 When GNAS R201H was introduced into a colorectal cancer cell line, increases in cyclic AMP and mucin production but not proliferation were observed.
A number of interesting findings appeared in the samples that underwent next-generation sequencing for PMP. The MCL1 and JUN mutations were novel for these patients, and they amplified concurrently in three patients. No additional single mutation for any individual tumor case was detected. Additionally, all three patients were long-term survivors. We hypothesize that the coamplification status of both MCL1 and JUN represents a new favorable prognostic marker in patients with PMP; however, a larger-scale analysis is required to determine this.
We also noticed that 2 of the 10 patients carried no detectable mutations by next-generation sequencing. However, at the time of analysis, patient 6 was alive with disease at 9.3 years of follow-up, while patient 7 died with disease 2.8 years after his diagnosis. It is possible that additional genetic aberrations harbored by their PMP tumors went undetected with the technique we chose for analysis. Certainly, more clinical, socioeconomic and pathological factors than genetic mutational status can influence a patient’s eventual survival.
Next-generation sequencing is a DNA-based technique, which requires further evaluation to validate its role in routine clinical practice. Amplifications of genes do not always correlate with gene expression, and other mechanisms can influence the level of protein expression. For example, next-generation sequencing may fail to capture epigenetic changes such as promoter methylation or histone acetylation that can also influence gene expression. Post translational events such as translational repression by microRNA are also not captured by next-generation sequencing. Although all cases in which we observed MCL1 amplification demonstrated some degree of MCL1 expression, expression of MCL1 was stronger in a few samples without amplification. The mechanisms of MCL1 expression in PMP are the subject of ongoing investigations.
Although our study is limited by the small sample size, the purpose of this research was to identify novel mutations that may help us in the treatment of PMP. Our clinical and gene-based results, especially regarding MCL1 and JUN amplification rates, are not epidemiological, as data were not controlled. Although PMP is rare, the patients affected by this disease need improved prognostication and therapies.
Conclusion
We identified coamplification of MCL1 and JUN in PMP. Additionally, we confirmed the numerous KRAS and GNAS mutations reported previously. MCL1 and JUN may represent novel prognostic markers or targets for treatment of PMP and require further study.
References
Smeenk, R. M., van Velthuysen, M. L., Verwaal, V. J. & Zoetmulder, F. A. Appendiceal neoplasms and pseudomyxoma peritonei: a population based study. Eur. J. Surg. Oncol. 34, 196–201 (2008).
Hinson, F. L. & Ambrose, N. S. Pseudomyxoma peritonei. Br. J. Surg. 85, 1332–1339 (1998).
Chua, T. C., Moran, B. J., Sugarbaker, P. H., Levine, E. A., Glehen, O., Gilly, F. N. et al. Early- and long-term outcome data of patients with pseudomyxoma peritonei from appendiceal origin treated by a strategy of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. J. Clin. Oncol. 30, 2449–2456 (2012).
Austin, F., Mavanur, A., Sathaiah, M., Steel, J., Lenzner, D., Ramalingam, L. et al. Aggressive management of peritoneal carcinomatosis from mucinous appendiceal neoplasms. Ann. Surg. Oncol. 19, 1386–1393 (2012).
Schomas, D. A., Miller, R. C., Donohue, J. H., Gill, S., Thurmes, P. J., Haddock, M. G. et al. Intraperitoneal treatment for peritoneal mucinous carcinomatosis of appendiceal origin after operative management: long-term follow-up of the Mayo Clinic experience. Ann. Surg. 249, 588–595 (2009).
Han, S. W., Kim, H. P., Shin, J. Y., Jeong, E. G., Lee, W. C., Lee, K. H. et al. Targeted sequencing of cancer-related genes in colorectal cancer using next-generation sequencing. PLoS One 8, e64271 (2013).
Chen, J., Ye, Y., Sun, H. & Shi, G. Association between KRAS codon 13 mutations and clinical response to anti-EGFR treatment in patients with metastatic colorectal cancer: results from a meta-analysis. Cancer Chemother. Pharmacol. 71, 265–272 (2013).
Al-Sohaily, S., Biankin, A., Leong, R., Kohonen-Corish, M. & Warusavitarne, J. Molecular pathways in colorectal cancer. J. Gastroenterol. Hepatol. 27, 1423–1431 (2012).
Ronnett, B. M., Zahn, C. M., Kurman, R. J., Kass, M. E., Sugarbaker, P. H. & Shmookler, B. M. Disseminated peritoneal adenomucinosis and peritoneal mucinous carcinomatosis: a clinicopathologic analysis of 109 cases with emphasis on distinguishing pathologic features, site of origin, prognosis, and relationship to ‘pseudomyxoma peritonei’. Am. J. Surg. Pathol. 19, 1390–1408 (1995).
Yelensky, R., Wang, K., Dogan, S., Borsu, L., Frampton, G., Lipson, D. et al. Next-generation sequencing of FFPE solid tumor specimens for clinical use. J Clin Oncol (Suppl), Abstract 10524 (2012).
Lipson, D., Capelletti, M., Yelensky, R., Otto, G., Parker, A., Jarosz, M. et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 18, 382–384 (2012).
Wilson, E. B. & Hilferty, M. M. The distribution of chi-square. Proc. Natl Acad. Sci. USA 17, 684–688 (1931).
Kaplan, E. L. & Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 53, 457–481 (1958).
Kozopas, K. M., Yang, T., Buchan, H. L., Zhou, P. & Craig, R. W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl Acad. Sci. U S A 90, 3516–3520 (1993).
Craig, R. W., Jabs, E. W., Zhou, P., Kozopas, K. M., Hawkins, A. L., Rochelle, J. M. et al. Human and mouse chromosomal mapping of the myeloid cell leukemia-1 gene: MCL1 maps to human chromosome 1q21, a region that is frequently altered in preneoplastic and neoplastic disease. Genomics 23, 457–463 (1994).
Moshynska, O., Sankaran, K., Pahwa, P. & Saxena, A. Prognostic significance of a short sequence insertion in the MCL-1 promoter in chronic lymphocytic leukemia. J. Natl Cancer Inst. 96, 673–682 (2004).
Schulze-Bergkamen, H., Fleischer, B., Schuchmann, M., Weber, A., Weinmann, A., Krammer, P. H. et al. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer 6, 232 (2006).
Ding, Q., He, X., Xia, W., Hsu, J. M., Chen, C. T., Li, L. Y. et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 67, 4564–4571 (2007).
Song, L., Coppola, D., Livingston, S., Cress, D. & Haura, E. B. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol. Ther. 4, 267–276 (2005).
Allen, T. D., Zhu, C. Q., Jones, K. D., Yanagawa, N., Tsao, M. S. & Bishop, J. M. Interaction between MYC and MCL1 in the genesis and outcome of non-small-cell lung cancer. Cancer Res. 71, 2212–2221 (2011).
Hogarty, M. D. Mcl1 becomes ubiquitin-ous: new opportunities to antagonize a pro-survival protein. Cell Res. 20, 391–393 (2010).
Graidist, P., Phongdara, A. & Fujise, K. Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5-fluorouracil-induced cytotoxicity. J. Biol. Chem. 279, 40868–40875 (2004).
Bohmann, D., Bos, T. J., Admon, A., Nishimura, T., Vogt, P. K. & Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 238, 1386–1392 (1987).
Haluska, F. G., Huebner, K., Isobe, M., Nishimura, T., Croce, C. M. & Vogt, P. K. Localization of the human JUN protooncogene to chromosome region 1p31-32. Proc. Natl Acad. Sci. USA 85, 2215–2218 (1988).
Hattori, K., Angel, P., Le Beau, M. M. & Karin, M. Structure and chromosomal localization of the functional intronless human JUN protooncogene. Proc. Natl Acad. Sci. USA 85, 9148–9152 (1988).
Wang, D. G., Johnston, C. F. & Buchanan, K. D. Oncogene expression in gastroenteropancreatic neuroendocrine tumors: implications for pathogenesis. Cancer 80, 668–675 (1997).
Volm, M., Drings, P. & Wodrich, W. Prognostic significance of the expression of c-fos, c-jun and c-erbB-1 oncogene products in human squamous cell lung carcinomas. J. Cancer Res. Clin. Oncol. 119, 507–510 (1993).
Shetty, S., Thomas, P., Ramanan, B., Sharma, P., Govindarajan, V. & Loggie, B. Kras mutations and p53 overexpression in pseudomyxoma peritonei: association with phenotype and prognosis. J. Surg. Res. 180, 97–103 (2013).
Kabbani, W., Houlihan, P. S., Luthra, R., Hamilton, S. R. & Rashid, A. Mucinous and nonmucinous appendiceal adenocarcinomas: different clinicopathological features but similar genetic alterations. Mod. Pathol. 15, 599–605 (2002).
Cuatrecasas, M., Matias-Guiu, X. & Prat, J. Synchronous mucinous tumors of the appendix and the ovary associated with pseudomyxoma peritonei: a clinicopathologic study of six cases with comparative analysis of c-Ki-ras mutations. Am. J. Surg. Pathol. (1996) 20, 739–746.
Szych, C., Staebler, A., Connolly, D. C., Wu, R., Cho, K. R. & Ronnett, B. M. Molecular genetic evidence supporting the clonality and appendiceal origin of pseudomyxoma peritonei in women. Am. J. Pathol. 154, 1849–1855 (1999).
Nash, G. M., Gimbel, M., Cohen, A. M., Zeng, Z. S., Ndubuisi, M. I., Nathanson, D. R. et al. KRAS mutation and microsatellite instability: two genetic markers of early tumor development that influence the prognosis of colorectal cancer. Ann. Surg. Oncol. 17, 416–424 (2010).
Andreyev, H. J., Norman, A. R., Cunningham, D., Oates, J., Dix, B. R., Iacopetta, B. J. et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br. J. Cancer 85, 692–696 (2001).
Ahnen, D. J., Feigl, P., Quan, G., Fenoglio-Preiser, C., Lovato, L. C., Bunn, P. A. Jr et al. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res. 58, 1149–1158 (1998).
Landis, C. A., Masters, S. B., Spada, A., Pace, A. M., Bourne, H. R. & Vallar, L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340, 692–696 (1989).
Lyons, J., Landis, C. A., Harsh, G., Vallar, L., Grunewald, K., Feichtinger, H. et al. Two G protein oncogenes in human endocrine tumors. Science 249, 655–659 (1990).
Nishikawa, G., Sekine, S., Ogawa, R., Matsubara, A., Mori, T., Taniguchi, H. et al. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br. J. Cancer 108, 951–958 (2013).
Acknowledgements
This work is supported by the Seed Grant for translational biology of pseudomyxoma peritonei, National Organization for Rare Disorders, BU70308/FP60037, awarded in June 2010. Portions of this work were presented in abstract form at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, USA, October 19–23, 2013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Sio, T., Mansfield, A., Grotz, T. et al. Concurrent MCL1 and JUN amplification in pseudomyxoma peritonei: a comprehensive genetic profiling and survival analysis. J Hum Genet 59, 124–128 (2014). https://doi.org/10.1038/jhg.2013.132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.132
Keywords
This article is cited by
-
Germline whole exome sequencing of a family with appendiceal mucinous tumours presenting with pseudomyxoma peritonei
BMC Cancer (2020)
-
The biological basis and function of GNAS mutation in pseudomyxoma peritonei: a review
Journal of Cancer Research and Clinical Oncology (2020)
-
The curious case of Gαs gain-of-function in neoplasia
BMC Cancer (2018)
-
A commentary on concurrent MCL1 and JUN amplification in pseudomyxoma peritonei: a comprehensive genetic profiling and survival analysis
Journal of Human Genetics (2014)
